
Drug Recalls 2021
Clobetasol Propionate Ointment USP, 0.05%, 60 g Tubes, Lot AC13786 Due to Microbial Contamination
Taro Pharmaceuticals has posted a lot recall of Clobetasol Propionate Ointment 0.05%, 60 g tubes.
About this recall:
Taro is voluntarily recalling one (1) lot of Clobetasol Propionate Ointment USP, 0.05% packaged in 60 gram tubes, to the consumer level. The product is being recalled due to microbial contamination. This recall only applies to tubes labeled with “Lot AC13786” and “Exp Dec 2022.”
What this means to you:
This lot is being recalled due to the presence of Ralstonia pickettii bacteria (R. pickettii), which was discovered by the manufacturer through routine testing. R. pickettii is present in the natural environment (soil, water) and for healthy individuals with intact skin, is unlikely to cause any localized or systemic infections. However, for individuals who are immunocompromised or whose skin is not intact (sunburn, psoriasis, abrasions), the potential exists that systemic infections may occur if the product is contaminated with R. pickettii. This is due to the presence of the steroid component which increases absorption of the ointment. If this bacterium circulates in the human blood stream it can cause life-threatening, invasive infections such sepsis, pneumonia, meningitis, inflammation of the bone or bone marrow, and infection in the joint fluid and joint tissues. To date, Taro has not received any adverse event reports related to this lot.
The product is used for the relief of inflammation and itching of the skin that is responsive to steroids. It is packaged in 60 gram aluminum tubes labeled with the name of the product, Clobetasol Propionate Ointment USP, 0.05% and the NDC # 51672-1259-03 (image of container label hyperlinked on the following page). The lot number and expiration date are displayed on the bottom of each tube and the end flap of the product carton.
Consumers with questions regarding this recall can contact Taro by calling 1-866-923-4914 or by e-mail at TaroPVUS@taro.com, Monday through Friday between 7 am and 7 pm, US Central Time. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
|
For more information regarding this FDA Recall Notification, please refer to the FDA website. FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then selecting prompt #2. |
Taro Issues Voluntary Nationwide Recall of Clobetasol Propionate Ointment USP, 0.05%, 60 g Tubes, Lot AC13786 Due to Microbial Contamination
FDA Publish Date: 12/30/2021
Taro Pharmaceuticals is voluntarily recalling one (1) lot of Clobetasol Propionate Ointment USP, 0.05% packaged in 60 g tubes to the consumer level. This recall ONLY applies to tubes labeled with “Lot AC13786” and “Exp Dec 2022”. No other lots of this product are impacted. This lot is being recalled due to the presence of Ralstonia pickettii bacteria (R. pickettii), which was discovered by the manufacturer through routine testing.
R. pickettii is present in the environment (soil, water) and for healthy individuals with intact skin, is unlikely to cause any localized or systemic infections. However, for individuals who are immunocompromised or whose skin is not intact (sunburn, psoriasis, abrasions), there is a reasonable possibility that systemic infections may occur if the product is contaminated with R. pickettii. This is due to the presence of the corticosteroid component which enhances absorption of the ointment. If this bacterium is circulating in the human blood stream it can cause life-threatening, invasive infections such sepsis, pneumonia, meningitis, inflammation of the bone or bone marrow, and infection in the joint fluid and joint tissues. To date, Taro has not received any adverse event reports related to this lot.
Lot # |
Amount |
Expiration Date |
|
AC13786 |
96 units |
December 2022 |
Clobetasol Propionate Ointment USP, 0.05% is indicated for the relief of inflammatory and pruritic manifestations of corticosteroid responsive dermatoses and is packaged in 60 g aluminum tubes with polypropylene puncture-tip caps. Each 60 g tube is labeled to indicate the name of the product, Clobetasol Propionate Ointment USP, 0.05% and the NDC # 51672-1259-03 (image of container label is linked on the following page). The lot number and expiration date are displayed on the bottom of each tube and the end flap of the product carton.
Ninety-six (96) units of lot AC13786 were distributed to two wholesale distributors in the United States (US) market between November 16 and December 6, 2021. These two wholesale distributors may have further distributed this lot to their retail customers for prescription dispensing to patients who were prescribed Clobetasol Propionate Ointment USP, 0.05%, 60 g.
Taro has notified the affected distributors by phone, e-mail, and letters via US Mail and is arranging for return of any containers of Clobetasol Propionate Ointment USP, 0.05%, 60 g, Lot AC13786 (expiration date of December 2022). Retail customers that have any quantities of Lot AC13786 which is being recalled, should stop distribution and return any unsold units to their wholesaler.
Consumers with questions regarding this recall can contact Taro by calling 1-866-923-4914 or by e-mail at TaroPVUS@taro.com, Monday through Friday between 7 am and 7 pm, US Central Time. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the US Food and Drug Administration.
Company Contact Information
Consumers:
- Taro Pharmaceuticals U.S.A., Medical Information
- 1-866-923-4914
- TaroPVUS@taro.com
Product Photos
Padagis has posted a lot recall of Nitroglycerin Lingual Spray 12 gram spray bottle.
About this recall:
Padagis has issued a voluntary nationwide recall to the consumer/user level of 3 lots of Nitroglycerin Lingual Spray (NDC 45802-0210-02). Out of an abundance of caution, this product is being recalled due to a complaint that the drug may not dispense from the spray bottle. This recall applies only to the 12 gram spray bottle and not the 4.9 gram spray bottle of this medication. The 3 specific recalled lots are listed in the table provided at the FDA press release on the following page.
What this means to you:
Nitroglycerin Lingual Spray is indicated for acute relief of an attack or prevention of chest pain due to coronary artery disease (CAD) in adult patients. If the product does not deliver the appropriate amount of nitroglycerin, the patient will likely continue to experience chest pain. The label advises that if relief is not obtained after 3 doses over 15 minutes, the patient should promptly seek medical attention. To date, Padagis has not received any reports of adverse events related to this recall.
Padagis is notifying its distributors/customers by express package delivery service as well as electronic mail and is arranging for return of all recalled products. All customers, healthcare providers, and consumers are instructed to examine their inventory for the recalled lots of Nitroglycerin Lingual Spray immediately and to quarantine, discontinue the distribution and use of, and return all recalled lots of the product. Patients who have this product should contact their healthcare provider for an alternate replacement before returning the recalled product.
Patients with questions regarding this recall can contact 888-266-7912 Monday to Friday from 8 am to 5 pm EST. Patients should contact their physician or healthcare provider if they have experienced any problems that may be related to using this product or any medical concerns.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online.
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then selecting prompt #2.
Padagis has issued a voluntary nationwide recall to the consumer/user level of 3 lots of Nitroglycerin Lingual Spray. Out of an abundance of caution, this product is being recalled due to a complaint that the drug may not dispense from the spray bottle. There is a remote risk that the product may not properly dispense medication to patients in the event of a malfunction of their dispensing unit. This recall applies only to the 12 gram spray bottle and not the 4.9 gram spray bottle of this medication. Lots recalled are listed in the table below.
|
Drug |
NDC |
Strength |
Net Contents |
Lot # |
Expiration |
|||
|
Nitroglycerin Lingual Spray |
45802-0210-02 |
400 mcg per spray |
12 grams |
150892
|
Oct 2022 Feb 2023
|
|||
Risk Statement: If the product does not deliver the appropriate amount of nitroglycerin, the patient will likely continue to experience chest pain. The label advises that if relief is not obtained after 3 doses over 15 minutes, the patient should promptly seek medical attention. To date, Padagis has not received any reports of adverse events related to this recall.
Nitroglycerin Lingual Spray is indicated for acute relief of an attack or prophylaxis of chest pain due to coronary artery disease (CAD) in adult patients. All packaging and branding on impacted units is that of Perrigo Company PLC. The product is packaged in a 12 gram bottle contained within a carton. The medication was distributed nationwide in the United States (US) to wholesalers and retailers.
Padagis is notifying its distributors and customers by express package delivery service as well as electronic mail and is arranging for return of all recalled products. All customers, healthcare providers (HCPs), and consumers are instructed to examine their inventory for recalled Nitroglycerin Lingual Spray immediately and to quarantine, discontinue the distribution and use of, and return all recalled lots of the product. Customers and HCPs are being provided recall information by Sedgwick Claims Management Services. All customers who have distributed this product to consumers have been requested to identify their customers and notify them immediately of the or 888-266-7912.
product recall.HCPs, distributors, and retailers that have product which is being recalled should stop distribution. Patients who have this product should contact their HCP for an alternate replacement before returning the recalled product. The necessary form to document product information, as well as other information, is available by contacting Sedgwick atpadagis5665@sedgwick.comor 888-266-7912.
Patients with questions regarding this recall can contact 888-266-7912 Monday to Friday from 8am to 5pm EST. Patients should contact their physician or HCP if they have experienced any problems that may be related to using this product or any medical concerns.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’sMedWatch Adverse EventReporting program either online, by regular mail or by fax.
- Complete and submit the report Online
- Regular Mail or Fax:Download form or call 1-800-332-1088to request a reporting form, then complete and return to the address on the pre-addressed form or submit by fax to1-800-FDA-0178
This recall is being conducted with the knowledge of the US Food and Drug Administration.
Company Contact Information
Consumers:
- Sedgwick888-266-7912
- padagis5665@sedgwick.com
Product Photos
- Click here for product photos.
Efficient Expands Voluntary Nationwide Recall to Consumers to Include 12 Additional Lots of Rompe Pecho CF, Rompe Pecho EX, Rompe Pecho MAX, and Rompe Pecho DM Sold in 2019 Due to Microbial Contamination Concerns
Efficient Laboratories has posted a lot recall of Rompe Pecho products.
About this recall:
Efficient Laboratories is expanding its voluntary nationwide recall to consumers to include an additional 12 lots of Rompe Pecho CF, Rompe Pecho EX, Rompe Pecho MAX, and Rompe Pecho DM due to microbial contamination concerns. These lots were distributed in 2019. To date, Efficient Laboratories has not received any reports of adverse events.
What this means to you:
In rare circumstances, consumption of these specific lots could result in illness. These products are used to treat symptoms of the flu and the common cold, and each are packaged in a box containing a bottle of the liquid product.
The lot numbers and expiration dates can be found on the bottom of the cartons. These Rompe Pecho products were distributed nationwide to wholesalers and retailers. Consumers that have Rompe Pecho EX, Rompe Pecho CF, Rompe Pecho DM, or Rompe Pecho MAX from the lots that are being recalled should stop using these products and discard them. The impacted 12 lots of Rompe Pecho product are listed in the chart on the following page.
Consumers with questions regarding this recall can contact Efficient Laboratories by phone at (305) 805-3456, Monday through Friday from 9 am to 4:30 pm EST. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
| Product Name |
Lots |
Expiration Date |
|
Rompe Pecho CF |
19F88 19G164 |
Jun 2022 Jul 2022 |
|
Rompe Pecho DM |
19F168 19G145 19G361 19G449 19G491 |
Jun 2022 Jul 2022 Jul 2022 Jul 2022 Jul 2022 |
|
Rompe Pecho EX |
19H20 19J98 19A418 19E411 |
Aug 2022 Sep 2022 Jan 2022 May 2022 |
|
Rompe Pecho MAX |
19G219 |
Jul 2022 |
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then selecting prompt #2.
Efficient Expands Voluntary Nationwide Recall to Consumers to Include 12 Additional Lots of Rompe Pecho CF, Rompe Pecho EX, Rompe Pecho MAX, and Rompe Pecho DM Sold in 2019 Due to Microbial Contamination Concerns
FDA Publish Date: 12/10/2021
Efficient Laboratories is expanding its voluntary nationwide recall to consumers to include an additional 12 lots of Rompe Pecho CF, Rompe Pecho EX, Rompe Pecho MAX, and Rompe Pecho DM due to microbial contamination concerns. These lots were distributed in 2019. To date, Efficient Laboratories has not received any reports of adverse events.
In rare circumstances, consumption of these specific lots could result in illness. These products are used to treat symptoms of the flu and the common cold, and each are packaged in a box containing a bottle of the liquid product. The impacted 12 lots of Rompe Pecho product are listed below in the chart:
|
Product Name |
Lots |
Expiration Date |
|
Rompe Pecho CF |
19F88 19G164 |
Jun 2022 Jul 2022 |
|
Rompe Pecho DM |
19F168 19G145 19G361 19G449 19G491 |
Jun 2022 Jul 2022 Jul 2022 Jul 2022 Jul 2022 |
|
Rompe Pecho EX |
19H20 19J98 19A418 19E411 |
Aug 2022 Sep 2022 Jan 2022 May 2022 |
|
Rompe Pecho MAX |
19G219 |
Jul 2022 |
The lot numbers and expiration dates can be found on the bottom of the cartons. These Rompe Pecho products were distributed nationwide to wholesalers and retailers.
Consumers that have Rompe Pecho EX, Rompe Pecho CF, Rompe Pecho DM, or Rompe Pecho MAX from these lots that are being recalled should stop using these products and discard them. Efficient Laboratories has notified its distributors of these lots. All distributors have confirmed there is no product in their inventory. In addition, a review of certain stores confirmed no inventory at the retail level as well.
Consumers with questions regarding this recall can contact Efficient Laboratories by phone at (305) 805-3456, Monday through Friday from 9 am to 4:30 pm EST. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Adverse events or product complaints experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online.
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
This recall is being conducted with the knowledge of the United States (US) Food and Drug Administration (FDA).
Link to FDA recall notification
Company Contact Information
Consumers:
- Efficient Laboratories
- 305-805-3456
Product Photos
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Sandoz has posted a lot recall of Enoxaparin Sodium Injection.
About this recall:
Sandoz is initiating a recall of 1 lot of Enoxaparin Sodium Injection, USP 40 mg/0.4 mL Single-Dose Syringes because a portion of the lot experienced a temperature excursion during shipment.
What this means to you:
The exposure to higher temperatures may have significantly impacted the recalled product’s (lot SAB06761A) effectiveness and thus there may be reasonable probability of risk for patients with health conditions that the product is intended to treat. These patients could be at risk for blood clots blocking blood vessels, an artery, or traveling to other tissues or organs causing pain, swelling, stroke, clots in the lung, or death as a result of the underlying condition. To date, Sandoz has not received any reports of adverse events or injuries related to this recall.
The product is used for prevention of deep vein thrombosis (DVT), a condition that occurs when a blood clot forms in a deep vein, usually in the legs, that can occur after surgeries or in patients with restricted mobility during illness. Enoxaparin is also used for prevention of complications associated with heart attacks.
Please note: this recall is specific to only one batch (SAB06761A) of Enoxaparin Sodium Injection, USP 40 mg/ 0.4 mL and does not apply to any other strengths of Sandoz Enoxaparin Sodium Injection, USP or to other lots of the 40 mg/0.4 mL. Any product returned that is not associated with this recall will be destroyed and no credit issued.
Consumers who have Enoxaparin Sodium Injection, USP 40 mg/0.4 mL (NDC 00781-3246-64 and lot number SAB06761A) which is being recalled, should stop taking the recalled product and immediately consult with their physician to attain another prescription. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Consumers should contact Sedgwick directly by phone at 844-265-7389 to return the recalled product. Representatives are available Monday to Friday from 8 am to 5 pm ET.
In case of any adverse reactions, please call Sandoz at (800) 525-8747 or email qa.drugsafety@sandoz.com. Customer service agents are available Monday to Friday from 8:30 am to 5 pm ET. Adverse events can also be reported to FDA online at www.fda.gov/medwatch/report.htm.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then selecting prompt #2.
Sandoz is initiating a recall of 1 lot (SAB06761A, Exp. 04/2023) of Enoxaparin Sodium Injection, USP 40 mg/0.4 mL Single-Dose Syringes to the consumer level. A portion of lot SAB06761A experienced a temperature excursion during shipment.
This lot was shipped to customers in the months of September and October 2021.
The exposure to higher temperatures may have significantly impacted the recalled product’s (lot SAB06761A) effectiveness and thus there may be reasonable probability of risk for patients with health conditions that the product is intended to treat.
These patients could be at risk for blood clots blocking blood vessels, an artery, or traveling to other tissues or organs causing pain, swelling, stroke, clots in the lung, or death as a result of the underlying condition. To date, Sandoz has not received any reports of adverse events or injuries related to this recall.
The product is used for prevention of deep vein thrombosis (DVT), a condition that occurs when a blood clot forms in a deep vein, usually in the legs, that can occur after surgeries or in patients with restricted mobility during illness. Enoxaparin is also used for prevention of complications associated with heart attacks. The product is packaged in cartons containing ten 0.4 mL syringes, NDC 00781-3246-64. Enoxaparin Sodium Injection was distributed nationwide in the United States (US) to wholesalers and retailers.
|
Product Name |
NDC Number |
Lot Number. |
Expiration Date. |
Date of Manufacture |
|
Enoxaparin Sodium Injection, |
00781-3246-64 |
SAB06761A |
04/2023 |
05/26/2021 |
Please note: this recall is specific to only one batch (SAB06761A) of Enoxaparin Sodium Injection, USP 40 mg/ 0.4 mL and does not apply to any other strengths of Sandoz Enoxaparin Sodium Injection, USP or to other lots of the 40 mg/0.4 mL SKU. Any product returned that is not associated with this recall will be destroyed and no credit issued.
Sandoz has already notified its wholesalers and retailers by mail and is arranging for return of all recalled product.
Consumers who have Enoxaparin Sodium Injection, USP 40 mg/0.4 mL (NDC 0781-3246-64 and lot number SAB06761A) which is being recalled, should stop taking the recalled product and immediately consult with their physician to attain another prescription. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Retailers and consumers should contact Sedgwick directly by phone at 844-265-7389 to return the recalled product. Representatives are available Monday to Friday, 8 am to 5 pm ET.
In case of any adverse reactions, please call Sandoz at (800) 525-8747 or email qa.drugsafety@sandoz.com. Customer service agents are available Monday to Friday from 8:30 am to 5 pm ET. Adverse events can also be reported to FDA online.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch
- Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report online
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form or submit by fax to 1-800-FDA-0178.
This recall is being conducted with the knowledge of the US Food and Drug Administration.
Link to FDA recall notification
Company Contact Information
Consumers:
- Sedgwick
- 844-265-7389 sandoz4623@sedgwick.com
Product Photos
Click here for product photos.
Date: 10/14/2021
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Lupin has posted a full lot recall of irbesartan tablets and irbesartan/hydrochlorothiazide (HCTZ) tablets.
About this recall:
Lupin is voluntarily recalling irbesartan tablets and irbesartan/HCTZ tablets to the consumer level. As part of Lupin’s ongoing assessment, analysis revealed that certain tested active pharmaceutical ingredient (API) batches, but not finished product batches, were above the limit for the impurity, N-nitrosoirbesartan. Although no reports of illness that appear to relate to this issue have been received, out of an abundance of caution, Lupin is recalling all batches of Irbesartan Tablets, USP 75 mg, 150 mg, and 300 mg as well as Irbesartan/HCTZ Tablets, USP 150 mg/12.5 mg and 300 mg/12.5 mg in the United States (US).
What this means to you:
N-nitrosoirbesartan impurity is a probable human carcinogen (a substance that could cause cancer) based on results from laboratory tests.
Irbesartan is an angiotensin II receptor blocker (ARB) indicated for treatment of hypertension (to lower blood pressure) and for the treatment of diabetic nephropathy in hypertensive patients with type 2 diabetes, an elevated serum creatinine, and proteinuria (protein in the urine).
Irbesartan/HCTZ tablets are a combination of irbesartan (an ARB) and HCTZ (a thiazide diuretic), indicated for hypertension in patients not adequately controlled with monotherapy or as an initial therapy in patients likely to need multiple drugs to achieve their blood pressure goals.
Patients taking irbesartan tablets, USP 75 mg, 150 mg, and 300 mg or irbesartan/HCTZ tablets, USP 150 mg/12.5 mg and 300 mg/12.5 mg that have been recalled are advised to continue taking their medication and contact their pharmacist, physician, or medical provider for advice regarding an alternative treatment. The link below provides a complete listing of recalled products.
Patients with questions regarding this recall should contact Inmar Rx Solutions at (855) 769-3988 / (855) 769-3989 Monday to Friday 9 am to 5 pm EST. For reimbursement, please have the recalled lots returned to Inmar Rx Solutions; the lot number can be found on the side of the bottle label.
|
For more information regarding this FDA Recall Notification, please refer to the FDA website. FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then selecting prompt #2. |
FDA Publish Date: 10/14/2021
Lupin is voluntarily recalling the below mentioned batches of irbesartan tablets and irbesartan/hydrochlorothiazide (HCTZ) tablets to the consumer level. As part of Lupin’s ongoing assessment, analysis revealed that certain tested active pharmaceutical ingredient (API) batches, but not finished product batches, were above the specification limit for the impurity, N-nitrosoirbesartan. Although no reports of illness that appear to relate to this issue have been received, out of an abundance of caution, Lupin is recalling all batches of Irbesartan Tablets, USP 75 mg, 150 mg, and 300 mg as well as Irbesartan/HCTZ Tablets, USP 150 mg/12.5 mg and 300 mg/12.5 mg in the United States (US).
Lupin discontinued the marketing of irbesartan and irbesartan/HCTZ tablets on January 7, 2021. The products were distributed nationwide in the US to wholesalers, drug chains, mail order pharmacies, and supermarkets.
Risk Statement: N-nitrosoirbesartan impurity is a probable human carcinogen (a substance that could cause cancer) based on results from laboratory tests.
From October 8, 2018 (the earliest date of shipment from the manufacturing site of any of the impacted batches), to September 30, 2021, Lupin received 4 reports of illness from irbesartan and 0 reports from irbesartan/HCTZ.
Irbesartan is an angiotensin II receptor blocker (ARB) indicated for treatment of hypertension (to lower blood pressure) and for the treatment of diabetic nephropathy in hypertensive patients with type 2 diabetes, an elevated serum creatinine, and proteinuria. Irbesartan Tablets, USP 75 mg, 150 mg, and 300 mg are packaged in 30- and 90-count bottles and were distributed nationwide in the US to wholesalers, drug chains, mail order pharmacies, and supermarkets.
The recalled lots are included in the table on the following page and have distribution dates from 10/20/2018 to 12/03/2020.
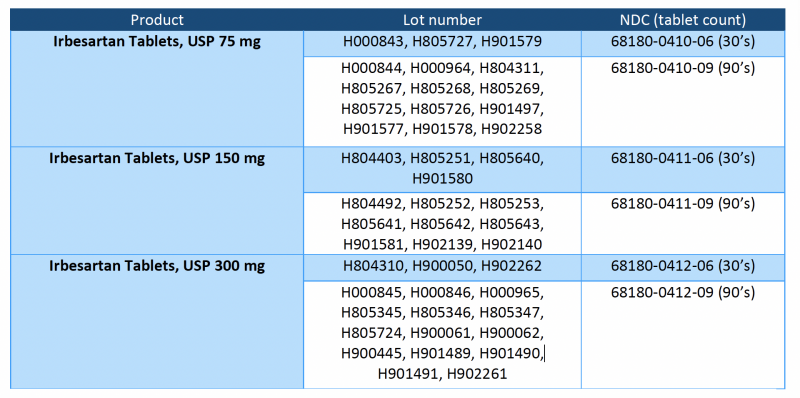
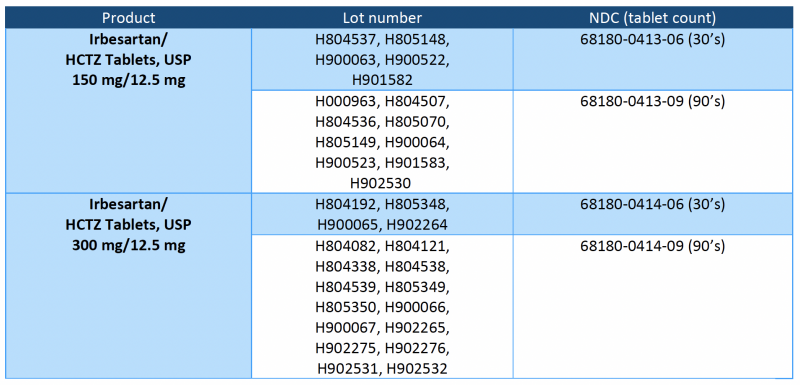
Irbesartan/HCTZ tablets are a combination of irbesartan (an ARB) and HCTZ (a thiazide diuretic), indicated for hypertension in patients not adequately controlled with monotherapy or as an initial therapy in patients likely to need multiple drugs to achieve their blood pressure goals. Irbesartan/HCTZ tablets, USP 150 mg/12.5 mg and 300 mg/12.5 mg are packaged in 30- and 90-count bottles. The recalled lots are included in the table below and have distribution dates from 10/17/2018 to 11/18/2020.
Lupin is notifying its wholesalers, distributors, drug chains, mail order pharmacies, and supermarkets by phone and through recall notification and is arranging for the return of all the recalled product lots.
Patients taking recalled irbesartan tablets, USP in the strengths of 75 mg, 150 mg, and 300 mg or irbesartan/HCTZ tablets USP, in the strengths of 150 mg/12.5 mg and 300 mg/12.5 mg are advised to continue taking their medication and contact their pharmacist, physician, or medical provider for advice regarding an alternative treatment.
Wholesalers, distributors, and retailers that have irbesartan and irbesartan/HCTZ tablets that are being recalled should discontinue distribution of the recalled product lots immediately and return it to Inmar Rx Solutions at 635 Vine Street, Winston Salem, NC 27101.
Consumers, wholesalers, distributors, and retailers with questions regarding this recall should contact Inmar Rx Solutions at (855) 769-3988 / (855) 769-3989 Monday to Friday 9 am to 5 pm EST. For reimbursement, please have the recalled lots returned to Inmar Rx Solutions; the lot number can be found on the side of the bottle label.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online.
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
This recall is being conducted with the knowledge of the US Food and Drug Administration.
Link to FDA recall notification
Company Contact Information
Consumers:
Inmar Rx Solutions, Inc.
(855) 769-3988 / (855) 769-3989
Media:
Shweta Munjal
shwetamunjal@lupin.com
Product Photos
Click here for product photos.
At Magellan Rx Management, we want to help you get the best possible care. We are contacting you to let you know that certain lots of Lotrimin® AF and Tinactin® Spray Products have been voluntarily recalled by the manufacturer.
About this recall:
Bayer is voluntarily recalling all unexpired Lotrimin® AF and Tinactin® spray products with lot numbers beginning with TN, CV, or NAA, distributed between September 2018 to September 2021, to the consumer level due to the presence of benzene in some samples of the products.
Drugs recalled (manufacturer):
Lotrimin AF and Tinactin Spray Products (Bayer)
NDCs impacted:
11523-1272-02, 11523-0010-02, 11523-4162-01, 11523-4140-02, 11523-0544-02, 11523-0777-02, 11523-0165-03, 11523-0072-05
What this means to you:
Benzene is classified as a human carcinogen. Exposure to benzene can occur by inhalation, orally, and through the skin. Depending on duration and level of exposure, it can result in cancers including leukemia as well as blood cancer of the bone marrow and blood disorders which can be life-threatening. Benzene is found in the environment from natural sources and human activity. Humans around the world are exposed to it from multiple sources and pathways, including inhalation, through the skin, and orally. To date, Bayer has no known reports of adverse events related to this recall. Bayer’s decision to voluntarily recall these products is a precautionary measure, and the levels detected are not expected to cause adverse health consequences in consumers.
The affected Lotrimin® and Tinactin® spray products are over-the-counter antifungal products, sold individually or in combo packs. Product images and information on which lot numbers fall under this recall are available at https://livewell.bayer.com/document/2011. There are no issues of concern with Lotrimin/Tinactin creams, including Lotrimin Ultra, or any other Bayer products. The recalled products are all packaged in aerosol spray cans. The products were distributed in the United States (US), Puerto Rico, Canada, and Mexico through a variety of retail channels.
Consumers may request a refund by visiting www.lotrimin.com or www.tinactin.com, and may contact Bayer with questions by calling 1-866-360-3266, Monday to Friday between the hours of 8 am and 8 pm Eastern Time. A photo of the product will be required to receive a refund. After taking your photo and completing the refund process, please discard the product appropriately. According to the FDA, consumers who have the products that are being recalled should stop using the product. Consumers should contact their healthcare provider if they have any questions, concerns or have experienced any problems related to using these aerosol antifungal products.
For more information regarding this FDA Recall Notification, please refer to the FDA website:
FDA contact information for reporting adverse events/quality complaints can be reached online at https://www.accessdata.fda.gov/scripts/medwatch/index.cfm?action=reporting.home or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then selecting prompt #2.
Reason for Recall:
Bayer is voluntarily recalling all unexpired Lotrimin® AF and Tinactin® spray products with lot numbers beginning with TN, CV, or NAA, distributed between September 2018 to September 2021, to the consumer level due to the presence of benzene in some samples of the products.
Drug recalled (manufacturer):
Lotrimin AF and Tinactin Spray Products (Bayer)
NDCs impacted:
11523-1272-02, 11523-0010-02, 11523-4162-01, 11523-4140-02, 11523-0544-02, 11523-0777-02, 11523-0165-03, 11523-0072-05
Actions for Member:
All members who have filled prescriptions through our systems for Lotrimin AF and Tinactin Spray Products in the past 3 months will also receive a written notice of this recall. The letter directs the member to discuss any concerns with their prescriber. You may be contacted by these members to discuss any impact. The summary below is being communicated to patients.
Benzene is classified as a human carcinogen. Exposure to benzene can occur by inhalation, orally, and through the skin. Depending on duration and level of exposure, it can result in cancers including leukemia as well as blood cancer of the bone marrow and blood disorders which can be life-threatening. Benzene is found in the environment from natural sources and human activity. Humans around the world are exposed to it from multiple sources and pathways, including inhalation, through the skin, and orally. To date, Bayer has no known reports of adverse events related to this recall. Bayer’s decision to voluntarily recall these products is a precautionary measure, and the levels detected are not expected to cause adverse health consequences in consumers.
The affected Lotrimin® and Tinactin® spray products are over-the-counter antifungal products, sold individually or in combo packs. Product images and information on which lot numbers fall under this recall are available at https://livewell.bayer.com/document/2011. There are no issues of concern with Lotrimin/Tinactin creams, including Lotrimin Ultra, or any other Bayer products. The recalled products are all packaged in aerosol spray cans. The products were distributed in the United States (US), Puerto Rico, Canada, and Mexico through a variety of retail channels.
Consumers may request a refund by visiting www.lotrimin.com or www.tinactin.com, and may contact Bayer with questions by calling 1-866-360-3266, Monday to Friday between the hours of 8 am and 8 pm Eastern Time. A photo of the product will be required to receive a refund. After taking your photo and completing the refund process, please discard the product appropriately. According to the FDA, consumers who have the products that are being recalled should stop using the product. Consumers should contact their healthcare provider if they have any questions, concerns or have experienced any problems related to using these aerosol antifungal products.
For more information regarding this FDA Recall Notification, please refer to the FDA website:
FDA contact information for reporting adverse events/quality complaints can be reached online at https://www.accessdata.fda.gov/scripts/medwatch/index.cfm?action=reporting.home or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then selecting prompt #2.
Eli Lilly Issues Voluntary Nationwide Recall of One Lot of Glucagon® Emergency Kit Due to Loss of Potency
Date: 09/26/2021
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Eli Lilly has posted a lot recall of Glucagon Emergency Kit.
About this recall:
Eli Lilly and Company (Lilly) is voluntarily recalling lot D239382D, expiration April 2022, of Glucagon Emergency Kit for Low Blood Sugar (glucagon for injection, 1 mg per vial; diluent for glucagon, 1 mL syringe), to the consumer (user) level. Lilly is recalling lot D239382D to the patient level because of a product complaint that the vial of glucagon was in liquid form instead of the powder form. The use of the liquid form of this product may fail to treat severe low blood sugar due to loss of potency.
Drug recalled (manufacturer): Glucagon Emergency Kit (Lilly)
NDC impacted (lot, expiration): 00002-8031-01 (lot: D239382D, expiration: April 2022)
What this means to you:
Severe hypoglycemia in patients with diabetes, if not reversed, can potentially cause adverse health consequences ranging from transient, minor complaints to neurological damage, seizures, and even death, if not promptly treated. Associated with the one product complaint, it was reported to Lilly that the involved patient experienced lack of drug effect and also reported subsequent seizures.
Glucagon Emergency Kit is used as an anti-hypoglycemic agent and a gastrointestinal motility inhibitor indicated for the treatment of severe hypoglycemia in pediatric and adult patients with diabetes mellitus. The product is packaged in a kit containing 1 mg of freeze-dried (lyophilized) product in a 3 mL vial and a pre-filled diluent syringe. The recalled Glucagon Emergency Kit, NDC 00002-8031-01, lot is D239382D, and the expiration date is April 2022 (label expiry date: 04 2022). The lot number can be found on the label of the kit as well as the vial.
Consumers in possession of Glucagon Emergency Kit lot D239382D should contact The Lilly Answers Center at 1-800-LILLYRX (1-800-545-5979) for return and replacement instructions for the product (Monday through Friday, 9 AM to 7 PM EST) and should contact their healthcare provider (HCP) for guidance. Consumers should contact their physician or HCP if they have experienced any problems that may be related to taking or using this product.
| For more information regarding this FDA Recall Notification, please refer to the FDA website:
FDA contact information for reporting adverse events/quality complaints can be reached online at https://www.accessdata.fda.gov/scripts/medwatch/index.cfm?action=reporting.home or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then selecting prompt #2. |
Eli Lilly Issues Voluntary Nationwide Recall of One Lot of Glucagon® Emergency Kit Due to Loss of Potency
FDA Publish Date: 09/26/2021
Eli Lilly and Company (Lilly) is voluntarily recalling lot D239382D, expiration April 2022, of Glucagon Emergency Kit for Low Blood Sugar (glucagon for injection, 1 mg per vial; diluent for glucagon, 1 mL syringe), to the consumer (user) level. Lilly is recalling lot D239382D to the patient level because of a product complaint that the vial of glucagon was in liquid form instead of the powder form. The firm’s investigation indicates that the liquid in this glucagon vial could be related to the manufacturing process. The use of the liquid form of this product may fail to treat severe low blood sugar due to loss of potency.
Risk Statement: Severe hypoglycemia in patients with diabetes, if not reversed, can potentially cause adverse health consequences ranging from transient, minor complaints to neurological damage, seizures, and even death, if not promptly treated. Associated with the one product complaint, it was reported to Lilly that the involved patient experienced lack of drug effect and also reported subsequent seizures.
Glucagon Emergency Kit is used as an anti-hypoglycemic agent and a gastrointestinal motility inhibitor indicated for the treatment of severe hypoglycemia in pediatric and adult patients with diabetes mellitus. The product is packaged in a kit containing 1 mg of freeze-dried (lyophilized) product in a 3 mL vial and a pre-filled diluent syringe. The recalled Glucagon Emergency Kit, NDC 00002-8031-01, lot is D239382D, and the expiration date is April 2022 (label expiry date: 04 2022). The lot number can be found on the label of the kit as well as the vial (refer to the photos linked below – Appendix A). The lot was distributed nationwide to wholesalers and retailers.
Lilly is notifying its distributors and customers by written communication and is arranging for return and replacement of all recalled products. Wholesalers and distributors with an existing inventory of Glucagon Emergency Kit lot D239382D should cease distribution and quarantine the product immediately.
Instructions for Wholesalers and Pharmacists
If you have distributed the recalled product, please notify any accounts or additional locations which may have received product from the recalled lot from you. Please conduct a sub-recall to those accounts and communicate this recall information immediately. Please request they immediately cease distribution of the product and promptly contact Sedgwick at 877-907-7032 (Interactive Voice Recording), 877-884-9410 (Fax), or elililly7484@sedgwick.com (Monday through Friday 8 am to 5 pm ET) to obtain a Business Reply Card (BRC) to initiate the return process.
Instructions for Consumers
Consumers in possession of Glucagon Emergency Kit lot D239382D should contact The Lilly Answers Center at 1-800-LILLYRX (1-800-545-5979) for return and replacement instructions for the product (Monday through Friday, 9 am to 7 pm EST) and should contact their healthcare provider (HCP) for guidance. Consumers should contact their physician or HCP if they have experienced any problems that may be related to taking or using this product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online.
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
This recall is being conducted with the knowledge of the United States Food and Drug Administration (FDA).
Link to FDA recall notification
Company Contact Information
Consumers:
The Lilly Answers Center
1-800-545-5979
Media:
Gregory A. Kueterman
317-432-5195
kueterman_gregory_andrew@lilly.com
Product Photos
Click here for product photos.
Pfizer Expands Voluntary Nationwide Recall to include All Lots of Chantix® (varenicline) Tablets Due to N-Nitroso-Varenicline
Date: 09/16/2021
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Pfizer has posted a full lot recall of Chantix (varenicline) 0.5 mg and 1 mg tablets.
About this recall:
Pfizer is voluntarily recalling all lots of Chantix 0.5 mg and 1 mg tablets to the patient (consumer/user) level due to the presence of a nitrosamine, N-nitroso-varenicline, at or above the FDA’s interim acceptable intake limit. This is an expansion of previously announced recalls.
Drug recalled (manufacturer): Chantix 0.5 mg and 1 mg tablets (Pfizer)
NDC(s) impacted: 0069-0468-56, 0069-0469-56, 0069-0469-03, 0069-0471-03
What this means to you:
Long-term ingestion of N-nitroso-varenicline may be associated with a potential increased cancer risk, but there is no immediate risk to patients taking this medication. The health benefits of stopping smoking outweigh the theoretical cancer risk from the nitrosamine impurity in varenicline. Nitrosamines are common in water and foods, including cured and grilled meats, dairy products, and vegetables. Everyone is exposed to some level of nitrosamines. These impurities may increase the risk of cancer if people are exposed to them above acceptable levels over long periods of time.
Chantix is a treatment to help patients quit smoking and is intended for short-term use. People who smoke cigarettes are 15 to 30 times more likely to get lung cancer than people who do not smoke. Smoking is also associated with many other cancers, as well as with heart and lung disease. Chantix has a safety profile that has been established over 15 years of marketing authorization. Pfizer believes the benefit/risk profile of Chantix remains positive. Patients currently taking Chantix should consult with their healthcare provider (HCP) about alternative treatment options. To date, Pfizer has not received reports of adverse events that have been related to this recall.
As communicated by the FDA, there is no immediate risk to patients taking Chantix. Patients who are taking this product should consult with their HCP to determine if alternative treatments are available. Patients with recalled Chantix tablets should contact Stericycle at 888-276-6166 (Monday to Friday, 8 am to 5 pm ET) for instructions on how to return their product and obtain reimbursement for their cost.
The NDCs, lot numbers, expiration dates, presentations, and photos for recalled Chantix tablets are available at the link provided on the following page.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online.
- Regular Mail or Fax: Download formor call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
| For more information regarding this FDA Recall Notification, please refer to the FDA website:
FDA contact information for reporting adverse events/quality complaints can be reached online at https://www.accessdata.fda.gov/scripts/medwatch/index.cfm?action=reporting.home or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then selecting prompt #2. |
Pfizer Expands Voluntary Nationwide Recall to include All Lots of Chantix® (varenicline) Tablets Due to N-Nitroso-Varenicline
FDA Publish Date: 09/16/2021
Pfizer is voluntarily recalling all lots of Chantix 0.5 mg and 1 mg tablets to the patient (consumer/user) level due to the presence of a nitrosamine, N-nitroso-varenicline, at or above the FDA’s interim acceptable intake limit. As alternative suppliers have been approved in the United States (US), Pfizer is undertaking this precautionary measure. This is an expansion of previously announced recalls.
Long-term ingestion of N-nitroso-varenicline may be associated with a theoretical increased cancer risk in humans, but there is no immediate risk to patients taking this medication. The health benefits from stopping smoking outweigh the potential cancer risk from the nitrosamine impurity in varenicline. Nitrosamines are common in water and foods, including cured and grilled meats, dairy products, and vegetables. Everyone is exposed to some level of nitrosamines. These impurities may increase the risk of cancer if people are exposed to them above acceptable levels over long periods of time.i
Chantix is a treatment to help patients quit smoking and is intended for short-term use. People who smoke cigarettes are 15 to 30 times more likely to get lung cancer than people who do not smoke.ii Smoking is also associated with many other cancers, as well as with cardiovascular disease and lung disease.iii Chantix has a safety profile that has been established over 15 years of marketing authorization and through a robust clinical program. Pfizer believes the benefit/risk profile of Chantix remains positive. Patients currently taking Chantix should consult with their healthcare professional (HCP) about alternative treatment options. To date, Pfizer has not received reports of adverse events assessed to be related to this recall.
Recalled product details are found in Appendix A on the following page. The lot numbers and photos for recalled products are provided at the link on the following page. The products were distributed nationwide to wholesalers and distributors in the US, US Virgin Islands, and Puerto Rico from May 2019 to September 2021. Pfizer has notified their direct consignees by letter to arrange for return of any recalled product. Wholesalers and distributors with an existing inventory of Chantix tablets, should stop use and distribution and quarantine the product immediately.
If you received free product through the Pfizer Patient Assistance Program (PAP) or the Pfizer Institutional Patient Assistance Program (IPAP), please check your stock immediately. If you have any of the product in inventory, please follow the instructions above for returning the product to Stericycle. Additionally, if you are aware of any patients to whom you dispensed the products and who still may have the product in their possession, please ask them to return the product to you and then follow the instructions above for returning the product to Stericycle. For any questions related to Pfizer PAP or Pfizer IPAP product, please contact 833-203- 2776 (Monday through Friday, 8 am to 6 pm ET).
As communicated by the FDA, there is no immediate risk to patients taking Chantix.iv Patients who are taking this product should consult with their HCP to determine if alternative treatments are available. Patients with Chantix tablets should contact Stericycle at 888-276-6166 (Monday through Friday, 8:00 am to 5:00 pm ET) for instructions on how to return their product and obtain reimbursement for their cost.
HCPs with questions regarding this recall can contact Pfizer using the below information.
| Contact Center |
Contact Information |
Area of Support |
|
Pfizer Medical Information |
800-438-1985, option 3 (Monday to Friday, 9 am to 5 pm ET) www.pfizermedinfo.com |
For medical questions regarding the product |
|
Pfizer Drug Safety |
800-438-1985, option 1 (24 hours a day; 7 days a week) |
To report adverse events and product complaints |
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online.
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
This recall is being executed with the knowledge of the US Food and Drug Administration (FDA).
Appendix A: Recalled Product Details
PRODUCT: Chantix Tablets, 0.5 mg NDC: 0069-0468-56 SIZE: Bottle of 56 Tablets EXPIRATION DATE: January 2022 – May 2023
PRODUCT: Chantix Tablets, 1 mg NDC: 0069-0469-56 SIZE: Bottle of 56 Tablets EXPIRATION DATE: September 2021 – December 2023
PRODUCT: Chantix Tablets, 1 mg NDC: 0069-0469-03 SIZE: Carton containing 4 blister packs of 14 tablets each EXPIRATION DATE: September 2021 – June 2023
PRODUCT: Chantix Tablets, 0.5/1 mg NDC: 0069-0471-03 SIZE: Carton containing 1 blister pack of eleven 0.5 mg tablets / 1 blister pack containing forty-two 1 mg tablets EXPIRATION DATE: August 2021 – January 2023
Link to FDA recall notification: https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts/pfizer-expands-voluntary-nationwide-recall-include-all-lots-chantixr-varenicline-tablets-due-n
3—0916-2021 Class 2 Chantix 0.5 mg and 1 mg Tablets – Pfizer’s Voluntary Nationwide Recall
Company Contact Information
Consumers:
Stericycle Inc.
888-276-6166
Media:
Eamonn Nolan
212-733-4626
Eamonn.Nolan@pfizer.com
Product Photos
Click here for product photos.
References
- https://www.fda.gov/drugs/drug-safety-and- availability/information-about-nitrosamine-impurities-medications
- US Centers for Disease Control and Prevention. What Are the Risk Factors for Lung Cancer? https://www.cdc.gov/cancer/lung/basic_info/risk_factors.htm Updated September 2020. Accessed June 2021.
- US Department of Health and Human Services. Smoking Cessation. A Report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health, 2020.
- https://www.fda.gov/drugs/drug-safety-and-availability/fda- alerts-health-care-professionals-and-patients-voluntary-recall- varenicline-chantix-warehouse
Jacobus Issues Voluntary Worldwide Recall of Ruzurgi® (amifampridine) 10 mg Tablets Due to Yeast, Mold, and Bacterial Contamination
Date: 09/13/2021
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Jacobus Pharmaceutical Company has posted a lot recall of Ruzurgi (amifampridine) 10 mg tablets.
About this recall:
Jacobus is voluntarily recalling 3 lots of Ruzurgi® (amifampridine) 10 mg tablets to the consumer level. The products have been found to be contaminated with yeast, mold, and aerobic bacteria based on laboratory test results.
What this means to you:
Oral products heavily contaminated with yeast, mold, and aerobic bacteria may result in serious and life-threatening infections. The use of the contaminated product in patients with underlying immunosuppressive conditions such as Lambert-Eaton myasthenic syndrome (LEMS) increases the potential for serious infections. Ruzurgi is used as a treatment for LEMS in patients ages 6 years old to less than 17 years old.
It is packaged in 100-count bottles (NDC: 49938-0110-01). The 3 recalled Ruzurgi lots include the following control numbers, expiration dates, and distribution dates:
|
Control Number* |
Expiration Date |
Distribution Dates |
|
18038 |
03/2023 |
05/25/2021 – 08/26/2021 (Canada only) |
|
18039 |
03/2023 |
06/01/2021 – 08/10/2021 |
|
18079 |
05/2023 |
08/10/2021 – 08/30/2021 |
* located to the right of the bottle’s front panel below the barcode
Jacobus is arranging for return of all recalled products. Consumers that have Ruzurgi which is being recalled should stop using and return the product.
If shipping via US Postal Service ship to:
Jacobus Pharmaceutical Company, Inc.
P.O. Box 5290
Princeton, NJ 08540
If shipping via courier service (e.g., UPS, FedEx) ship to: J
acobus Pharmaceutical Company, Inc.
IRL Building
31 Schalks Crossing Road
Plainsboro, NJ 08356
Consumers with questions regarding this recall can contact Jacobus by phone at (609) 799-8221 ext. 2120, Monday through Friday from 9 AM to 5 PM Eastern Standard Time. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
For more information regarding this FDA Recall Notification, please refer to the FDA website
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then selecting prompt #2.
acobus Issues Voluntary Worldwide Recall of Ruzurgi® (amifampridine) 10 mg Tablets Due to Yeast, Mold, and Bacterial Contamination
FDA Publish Date: 09/13/2021
Jacobus is voluntarily recalling 3 lots of Ruzurgi® (amifampridine) 10 mg tablets to the consumer level. The products have been found to be contaminated with yeast, mold, and aerobic bacteria based on laboratory test results.
Oral products heavily contaminated with yeast, mold, and aerobic bacteria may result in serious and life-threatening infections. The use of the contaminated product in patients with underlying immunosuppressive conditions such as Lambert-Eaton myasthenic syndrome (LEMS) increases the potential for serious infections. Ruzurgi is used as a treatment for LEMS in patients ages 6 years old to less than 17 years old.
It is packaged in 100-count bottles (NDC: 49938-0110-01). The 3 recalled Ruzurgi lots include the following control numbers, expiration dates, and distribution dates:
|
Control Number* |
Expiration Date |
Distribution Dates |
|
18038 |
03/2023 |
05/25/2021 – 08/26/2021 (Canada only) |
|
18039 |
03/2023 |
06/01/2021 – 08/10/2021 |
|
18079 |
05/2023 |
08/10/2021 – 08/30/2021 |
* located to the right of the bottle’s front panel below the barcode
Ruzurgi was distributed worldwide to specialty pharmacies and physicians. Jacobus was informed of this issue by their Canadian partner that was conducting confirmatory full testing on control number 18038. Jacobus conducted an expanded investigation which identified control numbers 18039 and 18079.
Jacobus is notifying its distributors and customers via regular mail and electronic mail and is arranging for return of all recalled products. Consumers that have Ruzurgi which is being recalled should stop using and return the product. Consumers with questions regarding this recall can contact Jacobus by phone at (609) 799-8221 ext. 2120, Monday through Friday from 9 AM to 5 PM Eastern Standard Time. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
If shipping via US Postal Service ship to:
Jacobus Pharmaceutical Company, Inc.
P.O. Box 5290
Princeton, NJ 08540
If shipping via courier service (e.g., UPS, FedEx) ship to:
Jacobus Pharmaceutical Company, Inc.
IRL Building
31 Schalks Crossing Road
Plainsboro, NJ 08356
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online.
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
This recall is being conducted with the knowledge of the United States (US) Food and Drug Administration (FDA) and other health authorities.
Company Contact Information
Consumers:
Jacobus Pharmaceutical Company Inc.
609-799-8221 ext. 2120
Product Photos
Click here for product photos
Azurity Issues Voluntary Nationwide Recall of One Lot of Firvanq® (Vancomycin Hydrochloride for Oral Solution), Vancomycin 50 mg/mL Kit, Due to a Mix-Up of the Diluent Included in the Kit
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Azurity has posted a lot recall of Firvanq® kit.
About this recall:
Azurity is voluntarily recalling one (1) lot of Firvanq (vancomycin hydrochloride [HCl] for oral solution), vancomycin 50 mg/mL kit to the consumer level, as some products in the recalled lot have been found to incorrectly contain a First® Omeprazole (FIRST®-PPI) diluent instead of the Firvanq diluent bottle.
- Drug recalled (manufacturer): Firvanq 50 mg/mL Kit (Azurity)
- NDC impacted (lot number): 65628-0206-05 (lot number: 21035)
What this means to you:
Vancomycin may not be completely dissolved in the FIRST-PPI diluent which could result in doses above or below those recommended. Taking inappropriate doses of oral vancomycin may potentially lead to persistent diarrhea associated with dehydration and electrolyte abnormalities, recurrence of Clostridium difficile (C. difficile) infection, progression to severe colitis, colon perforation requiring surgery, and potentially death. Especially, the elderly and immunocompromised patients are vulnerable to the complications of C. difficile infection. To date, Azurity has not received any reports of adverse events related to this recall.
Firvanq is indicated for use in adults and pediatric patients less than 18 years of age for the treatment of: Clostridium difficile-associated diarrhea and enterocolitis caused by Staphylococcus aureus (including methicillin-resistant strains). It is packaged as a kit consisting of a bottle with vancomycin HCl powder and a bottle of grape-flavored diluent. The product NDC is 65628-0206-05. A total of 2,751 kits of the recalled Firvanq, lot number 21035, with an expiration date of 07-31-2022 were distributed. The product can be identified by its brand name Firvanq, NDC, and lot number.
Azurity is arranging for return and replacement of only recalled products. Consumers that are in possession of Firvanq from the recalled lot should immediately stop using it and return it to the place of purchase.
Consumers with questions regarding this recall may contact Koral Couch, Senior Manager, Customer Service by phone: 781-935-8141 x 119; fax: 781 935-8395, or email: kcouch@azurity.com. Azurity business hours are Monday through Friday from 8:30 am to 5 pm Eastern Time. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
| For more information regarding this FDA Recall Notification, please refer to the FDA website
FDA contact information for reporting adverse events/quality complaints can be reached online at or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then selecting prompt #2. |
Azurity Issues Voluntary Nationwide Recall of One Lot of Firvanq® (Vancomycin Hydrochloride for Oral Solution), Vancomycin 50 mg/mL Kit, Due to a Mix-Up of the Diluent Included in the Kit
FDA Publish Date: 9/8/2021
Azurity is voluntarily recalling one (1) lot of Firvanq® (vancomycin hydrochloride [HCl] for oral solution), vancomycin 50 mg/mL kit to the consumer level, as some products in the recalled lot have been found to incorrectly contain a First® Omeprazole (FIRST®-PPI) diluent instead of the Firvanq diluent bottle.
Risk Statement: Vancomycin may not be completely solubilized in the FIRST-PPI diluent which could lead to doses above or below those recommended in the label. There is reasonable probability that the administration of inappropriate doses of oral vancomycin may lead to persistent diarrhea associated with dehydration and electrolyte abnormalities, recurrence of Clostridium difficile (C. difficile) infection, progression to severe colitis, colon perforation requiring colectomy, and potentially death. Especially, the elderly and immunocompromised patients are vulnerable to the complications of C. difficile infection. To date, Azurity has not received any reports of adverse events related to this recall.
Firvanq is indicated for use in adults and pediatric patients less than 18 years of age for the treatment of: Clostridium difficile-associated diarrhea and enterocolitis caused by Staphylococcus aureus (including methicillin-resistant strains). It is packaged as a kit consisting of a bottle with Vancomycin HCl, USP powder, a bottle of grape-flavored diluent, and the Prescribing Information. The product NDC is 65628-206-05 and the UPC code is 3 65628 206005 1. A total of 2,751 kits of the recalled Firvanq, lot number 21035, with an expiration date of 07-31-2022 were distributed. The product can be identified by its brand name Firvanq, NDC, and lot number. Product was distributed nationwide through wholesale distributors.
Azurity is notifying its distributors and customers by direct notifications to distributors and via this press release. Azurity is arranging for return and replacement of only recalled products. Consumers, distributors, and retailers that are in possession of Firvanq from the impacted lot should immediately stop using it and return it to the place of purchase. Distributors are asked to place any impacted product under quarantine and return it promptly to Azurity.
Consumers with questions regarding this recall may contact Koral Couch, Senior Manager, Customer Service by phone: 781-935-8141 x 119; fax: 781 935-8395, or email: kcouch@azurity.com. Azurity business hours are Monday through Friday from 8:30 am to 5 pm Eastern Time. Consumers should contact their physician or healthcare provider (HCP) if they have experienced any problems that may be related to taking or using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online.
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
This recall is being conducted with the knowledge of the United States (US) Food and Drug Administration (FDA).
Link to FDA recall notification
Company Contact Information
Consumer:
Koral Couch
781-935-8141 x 119
kcouch@azurity.com
Product Photos
Click here for product photos.
KVK Tech Issues Voluntary Nationwide Recall of Atovaquone Oral Suspension, USP 750 mg/5 mL Due to Temperature Abuse
Date: 08/06/2021
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
KVK Tech has posted a lot recall of Atovaquone Oral Suspension 750 mg/5 mL.
About this recall:
KVK Tech is voluntarily recalling 2 lots of Atovaquone Oral Suspension, USP 750 mg/5 mL to the consumer level. The recall is based on customer complaints of unusual grittiness in the product, which KVK has determined was most probably caused by prolonged exposure of these product lots to extremely cold weather during shipment.
What this means to you:
Atovaquone is a prescription drug used to treat Pneumocystis jiroveci [Pneumocystis carinii] pneumonia, a type of pneumonia most likely to affect people with human immunodeficiency virus (HIV) in teenagers and adults, and is also used to prevent patients with weakened immune systems from contracting this type of pneumonia.
Exposure of atovaquone oral suspension to extremely low temperatures may result in changes to the effectiveness, appearance, taste, and thickness of the liquid. As the product is required to be protected from freezing temperatures, severely immunocompromised patients who receive less effective drug may not have their serious or life-threatening infection treated adequately. To date, KVK Tech is not aware of any adverse events associated with this problem.
The product is packaged in 8 oz. bottles (210 mL bottle with child-resistant cap) packaged in a carton with NDC # 10702-223-21; the affected lots are labeled 16653A and 16654A with both lots having expiration dates of December 2022. The lot numbers and expiration dates can be found on the right bottom side of the labels on the bottles.
Patients or caregivers who have bottles of recalled atovaquone should stop using and should return the product to KVK Tech at 110 Terry Drive, Newtown, PA 18940. KVK Tech will arrange to reimburse customers for their costs in purchasing the product.
Consumers with questions regarding this recall can contact KVK Tech at recall@kvktech.com215-579-1842 Ext: 6002, Monday through Friday, 8 am to 4:30 pm EST or via email at . Consumers should contact their physician or healthcare professional if they have experienced any problems that may be related to taking or using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the United States (US) Food and Drug Administration. For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online.
KVK Tech Issues Voluntary Nationwide Recall of Atovaquone Oral Suspension, USP 750_mg/5 mL Due to Temperature Abuse
FDA Publish Date: 08/06/2021
KVK Tech is voluntarily recalling 2 lots of Atovaquone Oral Suspension, USP 750 mg/5 mL to the consumer level. The recall is based on customer complaints of unusual grittiness in the product, which KVK has determined was most probably caused by prolonged exposure of these product lots to extremely cold weather during shipment.
Exposure of atovaquone oral suspension to extremely low temperatures may result in changes to the effectiveness, appearance, taste, and thickness of the liquid. As the product is required to be protected from freezing temperatures, severely immunocompromised patients who receive less effective drug may experience inadequate treatment of serious and life-threatening infections. To date, KVK Tech is not aware of any adverse events associated with this problem.
Atovaquone is a prescription drug labeled to treat Pneumocystis jiroveci [Pneumocystis carinii] pneumonia, a type of pneumonia most likely to affect people with human immunodeficiency virus (HIV) in teenagers and adults, and is also used to prevent immunocompromised patients from contracting this type of pneumonia.
The product is packaged in 8 oz. bottles (210 mL bottle with child-resistant cap) packaged in a carton with NDC # 10702-223-21; the affected lots are labeled 16653A and 16654A, with both lots having expiration dates of December 2022. The lot numbers and expiration dates can be found on the right bottom side of the labels on the bottles.
The 2 lots of product were shipped to a single distributor, which has been notified as part of the recall. KVK Tech will be working with the distributor to ensure that the distributor’s customers return remaining inventory of the affected lots to KVK Tech for appropriate disposition. Patients or caregivers who have bottles of atovaquone affected by this recall should stop using and are requested to return the product to KVK Tech at 110 Terry Drive, Newtown, PA 18940, and KVK Tech will arrange to reimburse customers for their costs in purchasing the product.
Consumers with questions regarding this recall can contact KVK Tech at recall@kvktech.com215-579-1842 ext: 6002, Monday through Friday, 8 am to 4:30 pm EST or via email to . Consumers should contact their physician or healthcare professional if they have experienced any problems that may be related to taking or using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1-800-332-1088 1-800-FDA-0178 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to
This recall is being conducted with the knowledge of the United States (US) Food and Drug Administration.
Link to FDA recall notification.
Company Contact Information
Consumers:
KVK Tech
- 215-579-1842 Ext: 6002
- recall@kvktech.com
Media:
- Susan DiCroce
- (215) 579-1842 Ext: 1260
Product Details
For a listing of product photos: click here.
Pfizer Issues a Voluntary Nationwide Recall for 12 Lots of Chantix® (varenicline) Tablets Due to N-nitroso-varenicline Content
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Pfizer has posted a lot recall of Chantix (varenicline) tablets.
About this recall:
Pfizer is voluntarily recalling 2 lots of Chantix 0.5 mg tablets, 2 lots of Chantix 1 mg tablets, and 8 lots of the Chantix kit of 0.5 mg/1 mg tablets to the patient (consumer/user) level due to the presence of a nitrosamine, N-nitroso-varenicline, above the Pfizer established Acceptable Daily Intake (ADI) level.
What this means to you:
Nitrosamines are common in water and foods, including cured and grilled meats, dairy products, and vegetables. Everyone is exposed to some level of nitrosamines. These impurities may increase the risk of cancer if people are exposed to them above acceptable levels over long periods of time.
Long-term ingestion of N-nitroso-varenicline may be associated with a theoretical potential increased cancer risk in humans, but there is no immediate risk to patients taking this medication. The health benefits of stopping smoking outweigh the theoretical potential cancer risk from the nitrosamine impurity in varenicline.
Chantix is a treatment to help patients quit smoking and is intended for short term use. People who smoke cigarettes are 15 to 30 times more likely to get lung cancer than people who do not smoke. Smoking is also associated with many other cancers. Chantix has a safety profile that has been established over 15 years of marketing authorization and through a robust clinical program. Pfizer believes the benefit/risk profile of Chantix remains positive. Patients currently taking Chantix should consult with their healthcare provider (HCP) to confirm if they received a recalled lot, and if appropriate, about alternative treatment options. To date, Pfizer has not received any reports of adverse events that have been related to this recall.
As communicated by the FDA, there is no immediate risk to patients taking Chantix. Patients who are taking this product should consult with their HCP or pharmacy to determine if they have received a recalled lot. Patients with the recalled lots should contact Stericycle at 888-276-6166 (Monday to Friday 8 am to 5 pm ET) for instructions on how to return their product and obtain reimbursement for their cost.
The NDC, lot number, expiration date, and presentation for recalled Chantix tablets are available at the link provided on the following page.
Pfizer’s Voluntary Nationwide Recall For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then selecting prompt #2.
Pfizer is voluntarily recalling 2 lots of Chantix 0.5 mg tablets, 2 lots of Chantix 1 mg tablets, and 8 lots of the Chantix kit of 0.5 mg/1 mg tablets to the patient (consumer/user) level due to the presence of a nitrosamine, N-nitroso-varenicline, above the company established Acceptable Daily Intake (ADI) level.
Long-term ingestion of N-nitroso-varenicline may be associated with a theoretical potential increased cancer risk in humans, but there is no immediate risk to patients taking this medication. The health benefits of stopping smoking outweigh the potential cancer risk from the nitrosamine impurity in varenicline. Nitrosamines are common in water and foods, including cured and grilled meats, dairy products, and vegetables. Everyone is exposed to some level of nitrosamines. These impurities may increase the risk of cancer if people are exposed to them above acceptable levels over long periods of time.i
Chantix is a treatment to help patients quit smoking and is intended for short-term use. People who smoke cigarettes are 15 to 30 times more likely to develop lung cancer than people who do not smoke.ii Smoking is also associated with many other cancers.iii Chantix has a safety profile that has been established over 15 years of marketing authorization and through a robust clinical program. Pfizer believes the benefit/risk profile of Chantix remains positive. Patients currently taking Chantix should consult with their healthcare provider (HCP) to confirm if they received an impacted lot, and if appropriate, about alternative treatment options. To date, Pfizer has not received any reports of adverse events that have been related to this recall.
As communicated by the FDA, there is no immediate risk to patients taking Chantix.iv Patients who are taking this product should consult with their HCP or pharmacy to determine if they have received a recalled lot. Patients with the recalled lots should contact Stericycle at 888-276-6166 (Monday to Friday 8 am to 5 pm ET) for instructions on how to return their product and obtain reimbursement for their cost.
Pfizer places the utmost emphasis on patient safety and product quality at every step in the manufacturing and supply chain process. Pfizer has notified their direct consignees by letter to arrange for return of any recalled product. Wholesalers and distributors with an existing inventory of the lots, listed in the table below, should stop use and distribution and quarantine the product immediately. If you have further distributed the recalled product, please notify any accounts or additional locations which may have received the recalled product from you. Please conduct a sub-recall to those accounts and communicate this recall information immediately.
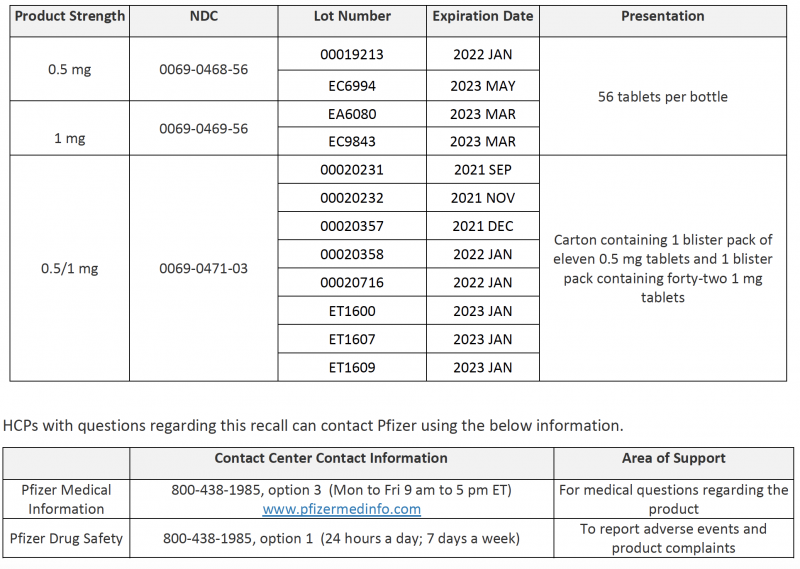
If you received free product through the Pfizer Patient Assistance Program (PAP) or the Pfizer Institutional Patient Assistance Program (IPAP), please check your stock immediately against the table below. If you have any of the affected product lots in your inventory, please follow the instructions on the press release for returning the product to Stericycle.
The NDC, lot number, expiration date, and presentation for recalled Chantix tablets are shown in the table below. The product lots were distributed nationwide to wholesalers and distributors in the United States (US) and Puerto Rico from June 2019 to June 2021.
Consumers with questions regarding this recall can contact the recall processor Eversana Life Science Services by phone at 1-888-304-5022, option 1; Monday to Friday, 8:00 am – 7:00 pm CDT. Consumers should contact their physician or HCP if they have experienced any problems that may be related to taking or using this drug product.
Customers with medical-related questions, who wish to report an adverse event, or quality issues about the recalled products should contact Viona by phone at 888-304-5011, Monday to Friday, 8:30 am – 5:30 pm EST.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
• Complete and submit the report Online
• Regular mail or fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the US FDA.
Link to FDA recall notification
Company Contact Information
Consumers:
Eversana Life Science Services
1-888-304-5022, option 1
Product Photos
Click here for product photos.
Viona Pharmaceuticals Issues Voluntary Nationwide Recall of Metformin HCl Extended-Release Tablets, 750 mg, Due to the Detection of NDMA Impurity
Date: 06/11/2021
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Viona Pharmaceuticals has posted a lot recall of Metformin HCl Extended-Release Tablets, USP 750 mg.
About this recall:
Viona is voluntarily recalling 2 lots of metformin hydrochloride (HCl) extended-release (ER) tablets, USP 750 mg to the retail level. The 2 lots of metformin HCl ER tablets, 750 mg have been found to contain levels of N-Nitrosodimethylamine (NDMA) above acceptable daily limits. This product was manufactured by Cadila Healthcare in November 2019 for United States (US) distribution by Viona.
What this means to you:
NDMA is classified as a substance that could cause cancer based on results from laboratory tests. NDMA is an environmental contaminant, found in water and foods (including meats, dairy products, and vegetables). Patients who have received recalled lots of metformin HCl ER tablets 750 mg are advised to continue taking their medication and contact their physician for advice regarding an alternative treatment. According to the FDA, it could be dangerous for patients with this serious condition to stop taking their metformin without first talking to their healthcare professionals (HCPs). To date, neither Viona, nor Cadila Healthcare have received any reports of adverse events related to this recall.
Metformin is used as an adjunct to diet and exercise to improve blood glucose control in adults with type 2 diabetes mellitus and is packaged in plastic bottles of 100 tablets, under NDC 72578-036-01. The recalled lots (batches) of metformin HCl ER tablets 750 mg are listed at the link in the box on the following page. The product can be identified as white to off-white, capsule shaped, uncoated tablets, debossed with “Z”, “C” on one side and “20” on the other side.
Consumers with questions regarding this recall can contact the recall processor Eversana Life Science Services by phone at 1-888-304-5022, option 1; Monday to Friday, 8:00 am – 7:00 pm CDT. Consumers should contact their physician or HCP if they have experienced any problems that may be related to taking or using this drug product.
Customers with medical-related questions, who wish to report an adverse event, or quality issues about the recalled products should contact Viona by phone at 888-304-5011, Monday to Friday, 8:30 am – 5:30 pm EST.
For more information regarding this FDA Recall Notification, please refer to the FDA website here.
FDA contact information for reporting adverse events/quality complaints can be reached online here or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then selecting prompt #2.
Viona Pharmaceuticals Issues Voluntary Nationwide Recall of Metformin HCl Extended-Release Tablets, USP 750 mg, Due to the Detection of NDMA Impurity
FDA Publish Date: 06/11/2021
Viona is voluntarily recalling 2 lots of metformin hydrochloride (HCl) extended-release (ER) tablets, USP 750 mg to the retail level. The 2 lots of metformin HCl ER tablets, USP 750 mg have been found to contain levels of N-Nitrosodimethylamine (NDMA) above acceptable daily limits. This product was manufactured by Cadila Healthcare in November 2019 for United States (US) distribution by Viona.
Risk Statement:
NDMA is classified as a probable human carcinogen (a substance that could cause cancer) based on results from laboratory tests. NDMA is a known environmental contaminant and is found in water and foods (including meats, dairy products, and vegetables). Patients who have received impacted lots of metformin HCl ER tablets, USP 750 mg are advised to continue taking their medication and contact their physician for advice regarding an alternative treatment. According to the Food and Drug Administration (FDA), it could be dangerous for patients with this serious condition to stop taking their metformin without first talking to their healthcare professionals (HCPs). Please visit the agency’s website for more information at https://www.fda.gov/drugs/drug-safety-and-availability/fda-updates-and-press-announcements-ndma-metformin. To date, neither Viona, nor Cadila Healthcare have received any reports of adverse events related to this recall.
The product is used as an adjunct to diet and exercise to improve blood glucose control in adults with type 2 diabetes mellitus and is packaged in high-density polyethylene (HDPE) plastic bottles of 100 tablets, under NDC 72578-036-01. The impacted lots of metformin HCl ER tablets, USP 750 mg are listed in the table on the following page. The product can be identified as white to off-white, capsule shaped, uncoated tablets, debossed with “Z”, “C” on one side and “20” on the other side. Product was distributed nationwide to distributors.

Viona is notifying its customers by email and mail (FedEx Overnight) and is arranging for return of all recalled products to the recall processor at the following address:
Eversana Life Science Services
c/o Viona recall
ATTN: Returns Department
4580 S. Mendenhall Rd.
Memphis, TN 38141
Consumers with questions regarding this recall can contact the recall processor Eversana Life Science Services by phone at 1-888-304-5022, option 1; Monday to Friday, 8:00 am – 7:00 pm CDT. Consumers should contact their physician or HCP if they have experienced any problems that may be related to taking or using this drug product.
Customers with medical-related questions, who wish to report an adverse event, or quality issues about the recalled products should contact Viona by phone at 888-304-5011, Monday to Friday, 8:30 am – 5:30 pm EST.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
• Complete and submit the report Online
• Regular mail or fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the US FDA.
Link to FDA recall notification: https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts/viona-pharmaceuticals-inc-issues-voluntary-nationwide-recall-metformin-hcl-extended-release-tablets
Company Contact Information
Consumers:
Eversana Life Science Services
1-888-304-5022, option 1
Product Photos
Click here for product photos.
Acella Issues Voluntary Nationwide Recall of Certain Lots of NP Thyroid® (Thyroid Tablets) Due to Sub Potency
Date: 04/30/2021
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Acella Pharmaceuticals has posted a lot recall of NP Thyroid® (thyroid tablets).
About this recall:
Acella is voluntarily recalling certain lots listed at the link on the following page of 15 mg, 30 mg, 60 mg, 90 mg, and 120 mg NP Thyroid (levothyroxine [T4] and liothyronine [T3]) to the consumer level. The products are being recalled as routine testing has found these lots to be subpotent. The product contains less than 90% of the labeled amount of liothyronine (T3) and/or levothyroxine (T4).
What this means to you:
The FDA is advising patients currently taking NP Thyroid from the recalled lots not to discontinue use without contacting their healthcare provider for further guidance and/or a replacement prescription.
Patients being treated for hypothyroidism (underactive thyroid) who receive subpotent NP Thyroid may experience signs and symptoms of hypothyroidism which may include fatigue, sensitivity to cold, constipation, dry skin, puffy face, hair loss, decreased heart rate, depression, thyroid gland swelling, and/or unexplained weight gain or difficulty losing weight. Pregnant women who receive subpotent product could experience miscarriage or negative fetal outcomes. In elderly patients and those with underlying cardiac disease, cardiac problems related to abnormal thyroid levels could occur.
Consumers with questions about the recall can email Acella at recall@acellapharma.com or contact Acella representatives at 1-888-424-4341, Monday through Friday from 8:00 am – 5:00 pm ET. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product. To best identify recalled product, the NDCs, product description, lot numbers, expiration dates, and product photos are listed at the link on the following page.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the United States (US) Food and Drug Administration (FDA).
For more information regarding this FDA Recall Notification, please refer to the FDA website. FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then selecting prompt #2.
Acella Issues Voluntary Nationwide Recall of Certain Lots of NP Thyroid® (Thyroid Tablets, USP) Due to Sub Potency
FDA Publish Date: 4/30/2021
Acella is voluntarily recalling certain lots listed at the link on the following page of 15 mg, 30 mg, 60 mg, 90 mg, and 120 mg NP Thyroid®, Thyroid Tablets, USP (levothyroxine [T4] and liothyronine [T3]) to the consumer level. The products are being recalled as routine testing has found these lots to be subpotent. The product contains less than 90% of the labeled amount of liothyronine (T3) and/or levothyroxine (T4).
Risk Statement: Patients being treated for hypothyroidism (underactive thyroid) who receive subpotent NP Thyroid may experience signs and symptoms of hypothyroidism which may include fatigue, sensitivity to cold, constipation, dry skin, puffy face, hair loss, decreased heart rate, depression, thyroid gland swelling, and/or unexplained weight gain or difficulty losing weight. There is reasonable risk of serious injury in newborn infants or pregnant women with hypothyroidism including the potential for early miscarriage, fetal thyroid problems, and/or impairments to fetal neural and skeletal development. In elderly patients and those with underlying cardiac disease, cardiac manifestations of thyroid problems may occur. To date, Acella has received 43 reports of serious adverse events that could possibly be related to this recall.
NP Thyroid is composed of levothyroxine and liothyronine and is used to treat hypothyroidism. The products subject to recall are packed in 100-count and 7-count bottles. Product images are provided at the link on the following page. To best identify recalled product, the NDCs, product description, lot numbers, and expiration dates are also listed at the link on the following page. These lots were distributed nationwide in the United States (US) to Acella’s direct accounts, including wholesalers, pharmacies, and healthcare offices.
Acella is proactively notifying its consignees to discontinue distribution of the recalled lots and is arranging for return of all recalled products. The FDA is advising patients currently taking NP Thyroid from the recalled lots not to discontinue use without contacting their healthcare provider for further guidance and/or a replacement prescription.
Consumers with questions about the recall can email Acella at recall@acellapharma.com or contact Acella representatives at 1-888-424-4341, Monday through Friday from 8:00 am – 5:00 pm ET. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the US Food and Drug Administration (FDA).
Link to FDA recall notification
Company Contact Information
Consumers:
Acella Pharmaceuticals
- 1-888-424-4341
- recall@acellapharma.com
Product Details
For a listing of NDCs, lot numbers, expiration dates, and product photos: click here.
Bryant Ranch Prepack Issues Voluntary Nationwide Recall of Spironolactone 25 mg and 50 mg Tablets Due to Mislabeling with the Incorrect Strength
Date: 3/9/2021
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Bryant Ranch Prepack has posted a lot recall of spironolactone tablets.
About this recall:
Bryant Ranch Prepack is voluntarily recalling in total 47 bottles of spironolactone tablets (4 different lots) to the consumer level. The products have been found to be mislabeled, displaying the incorrect strength. Prepackaged bottles labeled spironolactone 50 mg may contain spironolactone 25 mg tablets and prepackaged bottles of spironolactone 25 mg may contain spironolactone 50 mg tablets.
What this means to you:
A patient who takes spironolactone 25 mg instead of the prescribed spironolactone 50 mg may have an increase in blood pressure or increase in swelling due to excess fluid (edema). It is possible that patients could have a decrease in potassium, if taking half of the expected dose, which could lead to low potassium levels or hypokalemia, a condition associated with heart rhythm disorders. Additionally, patients who take spironolactone 50 mg instead of the prescribed spironolactone 25 mg could have an increase in potassium, which could be life threatening. Patients with decreased kidney function or those taking certain other medications (for example: RAAS inhibitors, such as lisinopril, captopril, and others) would be at increased risk. Consumers with questions regarding this recall can contact Bryant Ranch Prepack at 877-885-0882 Monday through Friday 6:30 am to 6 pm PST.
Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online.
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
For more information regarding this FDA Recall Notification, please refer to the FDA website. FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Bryant Ranch Prepack Issues Voluntary Nationwide Recall of Spironolactone 25 mg and 50 mg Tablets Due to Mislabeling with the Incorrect Strength
FDA Publish Date: 3/9/2021
Bryant Ranch Prepack is voluntarily recalling in total 47 bottles of spironolactone tablets (4 different lots) to the consumer level. The products have been found to be mislabeled, displaying the incorrect strength. Prepackaged bottles labeled spironolactone 50 mg may contain spironolactone 25 mg tablets and prepackaged bottles of spironolactone 25 mg may contain spironolactone 50 mg tablets.
A patient who consumes spironolactone 25 mg instead of the prescribed spironolactone 50 mg may experience an elevation in blood pressure or increased swelling caused by excess fluid (edema) if taking the product chronically. It is possible that patients could experience a decrease in potassium if taking half of the expected dose which could lead to hypokalemia, a condition associated with cardiac arrhythmias. Furthermore, patients who consume spironolactone 50 mg instead of the prescribed spironolactone 25 mg could experience an increase in potassium which could be life-threatening. Patients with renal insufficiency or those taking concomitant reninangiotensin-aldosterone system (RAAS) inhibitors would be at increased risk. As of 3/9/2021, Bryant Ranch Prepack has not received any reports of adverse events related to this recall.
Spironolactone is indicated as a diuretic in the treatment of high blood pressure, heart failure, hypokalemia, and edema and is repackaged in 30, 60 and 90-count bottles. Lots included in recall:
The product can be identified by the following details on the label: medication name as listed above with strength in a bold black box and a red and blue “BRP Pharmaceuticals” logo. Photos of the labels are provided in the photo section linked below.
Bryant Ranch Prepack is notifying its distributors and customers by mail and is arranging for return of all recalled products. Distributors that have existing inventory of any of the lots listed in this recall should contact Bryant Ranch Prepack immediately.
Consumers with questions regarding this recall can contact Bryant Ranch Prepack at 877-885-0882 Monday through Friday 6:30 am to 6 pm PST. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online.
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
This recall is being conducted with the knowledge of the United States (US) Food and Drug Administration (FDA).
Link to FDA recall notification
Company Contact Information Consumers:
- Bryant Ranch Prepack
- 877-885-0882
Product Photos: Click here for product photos.
Apotex Issues Nationwide Recall of Enoxaparin Sodium Injection Due to Mislabeling of Syringe Barrel Measurement Markings
Date: 2/3/2021
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information. Apotex has posted a lot recall of Enoxaparin Sodium Injection.
About this Recall:
Apotex is voluntarily recalling 2 batches of Enoxaparin Sodium Injection, USP to consumer level due to a packaging error resulting in some syringes barrels containing 150 mg/mL markings (corresponding to the 120 mg/0.8 mL strength) instead of 100 mg/mL markings (corresponding to the 100 mg/mL strength) on the syringe barrel and vice versa.
What this means to you:
Incorrect syringe barrel marking could result in inaccurate dose administration. In recalled batch CS008 (strength 100 mg/mL), patients could receive 3.75 mg of enoxaparin, instead of 3 mg of enoxaparin. In the other recalled batch (batch CT003, strength 120 mg/0.8 mL), patients could receive 2 mg of enoxaparin rather than 2.5 mg of enoxaparin. Accidentally taking too much drug may result in bleeding complications. Alternatively, if the dose administered is less than prescribed, blood clotting events could occur.
Enoxaparin sodium injection is used for the treatment of acute deep vein thrombosis (DVT), prevention of DVT which can lead to pulmonary embolism (PE), as well as for the prevention and treatment of certain cardiac events.
The recalled enoxaparin sodium injection can be identified by NDC numbers found on the carton and label of the product. The link on the following page provides these NDC numbers and additional details including the 2 recalled batch numbers.
Patients who have received either of the 2 impacted batches of enoxaparin sodium injection or have questions regarding this recall should contact their pharmacy. The FDA recommends patients not interrupt their therapy, immediately contact their healthcare provider for medical advice, and return the impacted product to Inmar Rx Solutions by contacting the number provided on the following page. 2— 0203-2021 Class 2 Enoxaparin Sodium Injection – Apotex Voluntary Nationwide Recall Consumers with the impacted units of Enoxaparin Sodium Injection, USP, please contact Inmar Rx Solutions (“Inmar”) at 1-855-667-8717, to receive a recall/return packet including the Recall Stock Response Form, or you may obtain this form from clsnetlink.com.
Consumers with questions regarding this recall can contact Apotex by phone at 1-800-706-5575 (8:30 am – 5 pm, EST Monday to Friday) or via email at UScustomerservice@Apotex.com.
Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Apotex Issues Voluntary Nationwide Recall of Enoxaparin Sodium Injection, USP Due to Mislabeling of Syringe Barrel Measurement Markings
FDA Publish Date: 2/3/2021
Apotex is voluntarily recalling 2 batches of Enoxaparin Sodium Injection, USP to the consumer level due to a packaging error resulting in some syringes barrels containing 150 mg/mL markings (corresponding to the 120 mg/ 0.8 mL strength) instead of 100 mg/mL markings (corresponding to the 100 mg/mL strength) on the syringe barrel and vice versa. The packaging error was discovered during a customer complaint investigation. To date, Apotex has not received any reports of adverse events related to use of these two batches. The affected product is manufactured by Gland Pharma.
Health Hazard Assessment:
Incorrect syringe barrel marking could lead to miscalculation and inaccurate dose administration. In one recalled batch (batch CS008, strength 100 mg/mL), if a consumer used a 150 mg/mL concentration packaged in a barrel corresponding to a 100 mg/mL concentration, the patient could receive 3.75 mg of enoxaparin, instead of 3 mg of enoxaparin. In another recalled batch (batch CT003, strength 120 mg/0.8 mL), if a consumer used a 100 mg/mL concentration packaged in a barrel corresponding to a 150 mg/mL concentration, the patient would receive 2 mg of enoxaparin rather than 2.5 mg of enoxaparin. Accidental overdosage could result in bleeding complications. Alternatively, if the dose administered is less than prescribed, the patient may experience blood clotting events.
Enoxaparin sodium injection is indicated for the prophylaxis of deep vein thrombosis (DVT) which may lead to pulmonary embolism (PE), treatment of acute DVT, prophylaxis of ischemic complications of unstable angina and non-Q-wave myocardial infarction (MI) when concurrently administered with aspirin, and treatment of acute STsegment elevation MI.
The 2 impacted batches of enoxaparin sodium injection, USP were distributed by Apotex nationwide in the United States (US) to wholesalers and warehousing chains. Apotex is currently notifying its affected direct account wholesalers and warehousing chains via mail (FedEx Standard Overnight) by mailing a recall notification letter and is arranging for return of all recalled product.
The impacted enoxaparin sodium injection can be identified by NDC numbers found on the carton and label of the product. The table on the following page also provides additional details including the 2 recalled batch numbers.
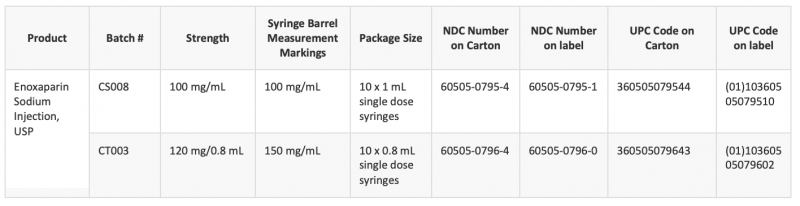
Patients who have received either of the 2 recalled batches of enoxaparin sodium injection or have questions regarding this recall should contact their pharmacy. The FDA recommends individuals not interrupt their therapy, immediately contact their healthcare provider for medical advice, and return the impacted product to Inmar Rx Solutions by contacting the numbers provided in this press release.
Consumers with the affected units of enoxaparin sodium injection, please contact Inmar Rx Solutions (“Inmar”) at 1-855-667-8717, to receive a recall/return packet including the Recall Stock Response Form, or you may obtain this form from clsnetlink.com.
Consumers with questions regarding this recall can contact Apotex by phone at 1-800-706-5575 (8:30 am – 5 pm, EST Monday to Friday) or via email at UScustomerservice@Apotex.com. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Wholesalers, distributors, and retailers should return the recalled product to the place of purchase. Anyone with existing inventory should quarantine the recalled batches immediately. Customers who purchased the impacted product directly from Apotex can call Inmar Rx Solutions at 1-855-667-8717 (9:00 am – 5:00 pm, EST Monday to Friday), to arrange for their return.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online.
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
This recall is being conducted with the knowledge of the US Food and Drug Administration.
Link to FDA recall notification
Company Contact Information Consumers: Apotex Corp.
- 1-800-706-5575
- UScustomerservice@Apotex.com
Media: Jordan Berman
- 1-416-749-9026
- jberman@apotex.com
Product Photos: Click here for product photos.
Nostrum Expands Voluntary Nationwide Recall of Metformin HCl Extended Release Tablets, USP 750 mg, Due to NDMA Content
Date: 1/25/2021
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Nostrum Laboratories has posted a recall of 1 lot of metformin hydrochloride (HCl) extended release (ER) tablets in the strength of 750 mg.
About this Recall:
Nostrum is voluntarily recalling one (1) lot of metformin HCl ER tablets in the strength of 750 mg to the consumer level. This product is the generic equivalent to Glucophage® tablets.
The recalled metformin HCl ER 750 mg tablets have been found to contain levels of nitrosamine impurities above the acceptable daily intake limit of 96 ng/day. This is an expansion of the recall initially announced on November 2, 2020.
What this means to you:
N-Nitrosodimethylamine (NDMA) is classified as a substance that could cause cancer based on results from lab tests. NDMA is a known environmental contaminant and is found in water and foods (including meats, dairy products, vegetables).
Metformin is used as an add-on to diet and exercise to improve blood glucose control in adults with type 2 diabetes mellitus. The recalled metformin ER 750 mg tablets are packaged in plastic bottles of 100 tablets with NDC 29033-056-01.
The recalled tablets carry lot number MET200601 with expiration date 07/2022. The product can be identified as an off-white oblong tablet debossed with “NM7”.
Consumers should consult a healthcare professional (HCP) to obtain a replacement or a different treatment option. It could be dangerous for patients with type 2 diabetes to stop taking their metformin without first talking to their HCP.
Consumers should contact their physician or HCP if they have experienced any problems that may be related to taking this drug product. Consumers with medical questions regarding this recall can contact Nostrum Medical Affairs at phone number 816-308-4941 or email quality@nostrumpharma.com Monday through Friday from 8 am – 5 pm CST. Consumers should contact their physician or pharmacy for further medical advice.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Nostrum Expands Voluntary Nationwide Recall of Metformin HCl Extended Release Tablets, USP 750 mg, Due to NDMA Content Above the Acceptable Daily Intake Limit
FDA Publish Date: 1/25/2021
Nostrum Laboratories is voluntarily recalling one (1) lot of metformin hydrochloride (HCl) extended release (ER) tablets, USP 750 mg (generic equivalent to Glucophage® tablets) to the consumer level. The metformin HCl ER tablets, USP 750 mg have been found to contain levels of nitrosamine impurities above the acceptable daily intake (ADI) limit of 96 ng/day as published in the Food and Drug Administration (FDA) Guidance Document issued September 2020. This is an expansion of the recall initially announced on November 2, 2020.
N-Nitrosodimethylamine (NDMA) is classified as a probable human carcinogen (a substance that could cause cancer) based on results from laboratory tests. NDMA is a known environmental contaminant and is found in water and foods (including meats, dairy products, vegetables). To date, Nostrum has not received any reports of adverse events related to this recall.
The product is indicated as an adjunct to diet and exercise to improve blood glucose control in adults with type 2 diabetes mellitus and is packaged in high density polyethylene (HDPE) bottles of 100 tablets, under NDC 29033- 056-01. The affected lot of metformin HCl ER tablets, USP 750 mg is listed in the table. The product can be identified as an off-white oblong tablet debossed with “NM7”. Metformin HCl ER tablets, USP 750 mg was distributed nationwide to wholesalers.
Product Description:
Metformin HCl Extended Release Tablets, USP 750 mg
NDC:
29033-056-01
Lot Number:
MET200601
Expiry Date:
07/2022
Nostrum is notifying its distributors by letter and is arranging for return of all recalled products. Pharmacies that have metformin HCl ER tablets, USP 750 mg which are being recalled should return to place of purchase. Consumers should consult a healthcare professional (HCP) to obtain a replacement or a different treatment option. It could be dangerous for patients with type 2 diabetes to stop taking their metformin without first talking to their HCP. Consumers should contact their physician or HCP if they have experienced any problems that may be related to taking this drug product.
Consumers with medical questions regarding this recall can contact Nostrum Medical Affairs by phone at 816-308- 4941 or email quality@nostrumpharma.com Monday through Friday from 8 am – 5 pm CST.
Consumers should contact their physician or pharmacy for further medical advice. Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online.
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
This recall is being conducted with the knowledge of the United States (US) FDA.
Link to FDA recall notification
Company Contact Information Consumers: Nostrum Laboratories, Inc.
- Medical Affairs 816-308-4941
- quality@nostrumpharma.com
Product Photos: Click here for product photos
Fresenius Kabi Issues Voluntary Nationwide Recall of Ketorolac Tromethamine Injection Due to the Presence of Particulate Matter
Date: 1/8/2021
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Fresenius Kabi has posted a lot recall of Ketorolac Tromethamine Injection, 30 mg/mL.
About this Recall:
Fresenius Kabi is voluntarily recalling a single lot of Ketorolac Tromethamine Injection, 30 mg/mL, in a 2 mL amber vial to the user level due to the presence of particulate matter.
What this means to you:
Ketorolac tromethamine, a nonsteroidal anti-inflammatory drug (NSAID), is indicated for the short-term (up to 5 days in adults) management of moderately severe acute pain that requires analgesia at the opioid level. Particulate matter was found in reserve sample vials. No adverse event reports have been received for the recalled lot. Administration of products containing particulate matter could obstruct blood vessels and result in local irritation of blood vessels, swelling at the site of injection, a mass of tissue that could become inflamed and infected, blood clots traveling to the lung, scarring of the lung tissues, and potentially life-threatening allergic reactions.
Customers with questions regarding this recall may contact Fresenius Kabi at 1-866-716-2459 Monday through Friday, during the hours of 8:00 a.m. to 5:00 p.m. Central Time. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product. The NDC, lot number, and expiration date for the recalled product are available at the link below.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Fresenius Kabi Issues Voluntary Nationwide Recall of Ketorolac Tromethamine Injection, Due to the Presence of Particulate Matter
FDA Publish Date: 1/8/2021
Fresenius Kabi is voluntarily recalling a single lot of Ketorolac Tromethamine Injection, USP, 30 mg/mL, 1 mL fill in a 2 mL amber vial to the user level due to the presence of particulate matter. Particulate matter was found in reserve sample vials. No adverse event reports have been received for the recalled lot, which was produced and sold in 2019.
Administration of products containing particulate matter could obstruct blood vessels and result in local irritation of blood vessels, swelling at the site of injection, a mass of tissue that could become inflamed and infected, blood clots traveling to the lung, scarring of the lung tissues, and potentially life-threatening allergic reactions.
Ketorolac tromethamine, a nonsteroidal anti-inflammatory drug (NSAID), is indicated for the short-term (up to 5 days in adults) management of moderately severe acute pain that requires analgesia at the opioid level. The total combined duration of use of oral ketorolac tromethamine and ketorolac tromethamine injection should not exceed 5 days.
Listed below is a table of the recalled lot distributed nationwide in the United States to wholesalers, distributors, hospitals, and pharmacies between March 28, 2019 and September 3, 2019. An image of the label is also included at the link on the following page. Product Name/Product size ND
Product Name/Product size:
Ketorolac Tromethamine Injection, USP, 30mg / mL, 1 mL fill in a 2 mL amber vial
NDC Number:
63323- 162-01
Product Code:
160201
Batch Number:
6121083
Expiration Date:
02/2021
First Ship Date:
03/28/2019
Last Ship Date:
09/03/2019
Fresenius Kabi is notifying its distributors and customers by letter and asking customers and distributors to check their stock immediately and to quarantine and discontinue the use and distribution of any affected product. Distributors should notify their customers and direct them to quarantine and discontinue distributing or 2—0108-2021 Class 1 Ketorolac Tromethamine Injection – Fresenius Kabi Voluntary Nationwide Recall dispensing the affected lot, and to return the product to Fresenius Kabi. The recall letter and response form are available at https://www.fresenius-kabi.com/us/pharmaceutical-product-updates.
Customers with questions regarding this recall may contact Fresenius Kabi at 1-866-716-2459 Monday through Friday, during the hours of 8:00 a.m. to 5:00 p.m. Central Time.
Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report online.
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
- Or, contact Fresenius Kabi at 1-800-551-7176, Monday through Friday, during the hours of 8:00 a.m. to 5:00 p.m. or via email at: productcomplaint.USA@fresenius-kabi.com or adverse.events.USA@freseniuskabi.com.
This recall is being conducted with the knowledge of the United States (US) Food and Drug Administration (FDA).
Link to FDA recall notification.
Company Contact Information Consumers: Fresenius Kabi
- 1-866-716-2459
Product Photos: Click here for product photos.
Nostrum Expands Voluntary Nationwide Recall of Metformin HCl Extended Release Tablets, USP 750 mg, Due to NDMA Content
Date: 1/4/2021
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Nostrum Laboratories has posted a recall of 1 lot of metformin hydrochloride (HCl) extended release (ER) tablets in the strength of 750 mg.
About this Recall:
Nostrum is voluntarily recalling one (1) lot of metformin HCl ER tablets in the strength of 750 mg to the consumer level. This product is the generic equivalent to Glucophage® tablets. The recalled metformin HCl ER 750 mg tablets have been found to contain levels of nitrosamine impurities above the acceptable daily intake limit of 96 ng/day. This is an expansion of the recall initially announced on November 2, 2020.
What this means to you:
N-Nitrosodimethylamine (NDMA) is classified as a substance that could cause cancer based on results from lab tests. NDMA is a known environmental contaminant and is found in water and foods (including meats, dairy products, vegetables).
Metformin is used as an add-on to diet and exercise to improve blood glucose control in adults with type 2 diabetes mellitus. The recalled metformin ER 750 mg tablets are packaged in plastic bottles of 100 tablets with NDC 29033-056-01. The recalled tablets carry lot number MET200501 with expiration date 07/2022. The product can be identified as an off-white oblong tablet debossed with “NM7”.
Consumers should consult a healthcare professional (HCP) to obtain a replacement or a different treatment option. It could be dangerous for patients with type 2 diabetes to stop taking their metformin without first talking to their HCP.
Consumers should contact their physician or HCP if they have experienced any problems that may be related to taking this drug product. Consumers with medical questions regarding this recall can contact Nostrum Medical Affairs at phone number 816-308-4941 or email quality@nostrumpharma.com Monday through Friday from 8 am – 5 pm CST. Consumers should contact their physician or pharmacy for further medical advice.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Nostrum Expands Voluntary Nationwide Recall of Metformin HCl Extended Release Tablets, USP 750 mg, Due to NDMA Content Above the Acceptable Daily Intake
Limit FDA Publish Date: 1/4/2021
Nostrum Laboratories is voluntarily recalling one (1) lot of metformin hydrochloride (HCl) extended release (ER) tablets, USP 750 mg (generic equivalent to Glucophage® tablets) to the consumer level. The metformin HCl ER tablets, USP 750 mg have been found to contain levels of nitrosamine impurities above the acceptable daily intake (ADI) limit of 96 ng/day as published in the Food and Drug Administration (FDA) Guidance Document issued September 2020. This is an expansion of the recall initially announced on November 2, 2020.
N-Nitrosodimethylamine (NDMA) is classified as a probable human carcinogen (a substance that could cause cancer) based on results from laboratory tests. NDMA is a known environmental contaminant and is found in water and foods (including meats, dairy products, vegetables). To date, Nostrum has not received any reports of adverse events related to this recall.
The product is indicated as an adjunct to diet and exercise to improve blood glucose control in adults with type 2 diabetes mellitus and is packaged in high density polyethylene (HDPE) bottles of 100 tablets, under NDC 29033- 056-01. The affected lot of metformin HCl ER tablets, USP 750 mg is listed in the table. The product can be identified as an off-white oblong tablet debossed with “NM7”. Metformin HCl ER tablets, USP 750 mg was distributed nationwide to wholesalers.
Product Description:
Metformin HCl Extended Release Tablets, USP 750 mg
NDC:
29033-056-01
Lot Number:
MET200501
Expiry Date:
07/2022
Nostrum is notifying its distributors by letter and is arranging for return of all recalled products. Pharmacies that have metformin HCl ER tablets, USP 750 mg which are being recalled should return to place of purchase. Consumers should consult a healthcare professional (HCP) to obtain a replacement or a different treatment option. It could be dangerous for patients with type 2 diabetes to stop taking their metformin without first talking to their HCP. Consumers should contact their physician or HCP if they have experienced any problems that may be related to taking this drug product.
Consumers with medical questions regarding this recall can contact Nostrum Medical Affairs by phone at 816-308- 4941 or email quality@nostrumpharma.com Monday through Friday from 8 am – 5 pm CST. Consumers should contact their physician or pharmacy for further medical advice.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online.
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
This recall is being conducted with the knowledge of the United States (US) FDA.
Link to FDA recall notification.
Company Contact Information Consumers: Nostrum Laboratories, Inc.
- Medical Affairs 816-308-4941
- quality@nostrumpharma.com
Product Photos: Click here for product photos.
Click here for language services (translation, TTY, etc.)