
Drug Recalls 2020
Sunstar Americas Expands Voluntary Nationwide Recall of Paroex® Chlorhexidine Gluconate Oral Rinse USP, 0.12% Due to Microbial Contamination
Date: 12/28/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information. Sunstar Americas has posted a market recall of Paroex® Chlorhexidine Gluconate Oral Rinse.
About this Recall:
Sunstar Americas, Inc. (SAI) is voluntarily recalling Paroex® Chlorhexidine Gluconate Oral Rinse USP, 0.12% products with an expiration date from 12/31/2020 – 9/30/2022 to the consumer level. The product may be contaminated with the bacteria Burkholderia lata. This is an expansion of the recall initially announced in October 2020.
What this means to you:
Use of the contaminated product may lead to a mouth infection and, potentially, an infection in the body that requires antibacterial treatment. In the most at-risk populations, the use of the contaminated product may lead to life-threatening infections, such as pneumonia and bacteremia.
Consumers with questions regarding this recall can contact SAI by phone at 1-800-528-8537 or email at us.pcr@us.sunstar.com Monday to Friday from 8 am to 5 pm CST. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to using this drug product.
For more information regarding this FDA Recall Notification, please refer to the FDA website. FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Sunstar Americas Expands Voluntary Nationwide Recall of Paroex® Chlorhexidine Gluconate Oral Rinse USP, 0.12% Due to Microbial Contamination FDA Publish
Date: 12/28/2020
Sunstar Americas, Inc. (SAI) is voluntarily recalling Paroex® Chlorhexidine Gluconate Oral Rinse USP, 0.12% products with an expiration date from 12/31/2020 – 9/30/2022 to the consumer level. The product may be contaminated with the bacteria Burkholderia lata. This is an expansion of the recall initially announced on October 27, 2020.
Use of the contaminated product in an immunocompetent host may lead to oral and, potentially, systemic infections requiring antibacterial therapy. In the most at-risk populations, the use of the contaminated product may result in life-threatening infections, such as pneumonia and bacteremia.
To date, 29 adverse events have been reported to the manufacturer related to this recall. Affected patients tested positive for Burkholderia lata infections, usually found in sputum cultures, while under treatment for other serious medical conditions. Use of the contaminated product in patients with pre-existing respiratory conditions, including in those with coronavirus disease 2019 (COVID-19), is particularly unsafe.
The prescription oral rinse product, available through healthcare professionals (HCPs) only, is indicated for use as part of a professional program for the treatment of gingivitis and is packaged as follows:
- 1789P GUM® Paroex® is distributed in cases each containing 6 amber bottles of 16 fluid ounce (473 mL) chlorhexidine rinse. The bottle has a childproof cap and a 15 mL metered dosage cup, is safety sealed, and is decorated with a multiple-panel wrap-around label.
- 1788P GUM® Paroex® is distributed in cases each containing 24 amber bottles of 4 fluid ounce (118.25 mL) chlorhexidine rinse. The bottle has a childproof cap, is safety sealed, and is decorated with a multiplepanel wrap-around label.
Paroex was distributed nationwide to dental offices, dental distributors, pharmaceutical wholesalers, dental schools, and pharmacies.
SAI is notifying its direct distributors and customers by USPS Priority mail and is arranging for return of all recalled products. Patients, pharmacies, and healthcare facilities in possession of these products should stop using and dispensing immediately. Sunstar is committed to delivering safe, fully compliant products of the highest quality and is taking necessary steps to prevent future occurrence of this issue.
Consumers with questions regarding this recall can contact SAI by phone at 1-800-528-8537 or email at us.pcr@us.sunstar.com Monday through Friday from 8 am to 5 pm CST. Consumers should contact their physician or HCP if they have experienced any problems that may be related to using this drug product.
Affected products and lot numbers follow below:
- Product name: Paroex® Chlorhexidine Gluconate Oral Rinse USP, 0.12%
- Size/ Form: 16 fl.oz. amber bottles
- NDC #: 052376-021-02
- Product Code: 1789P
- Lots Recalled: all lots with expiration date from December 31, 2020 through September 30, 2022
Product name: Paroex® Chlorhexidine Gluconate Oral Rinse USP, 0.12% o Size/ Form: 4 fl.oz. Amber Bottles o NDC #: 052376-021-04 o Product Code: 1788P o Lots Recalled: all lots with expiration date from December 31, 2020 through September 30, 2022 Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online.
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
This recall is being conducted with the knowledge of the United States (US) Food and Drug Administration (FDA).
Link to FDA recall notification.
Company Contact Information Consumers: Sunstar Americas
- 1-800-528-8537
- us.pcr@us.sunstar.com
Media: Greg Belair
- 847-794-4241
- Greg.belair@us.sunstar.com
Product Photos: Click here for product photos.
Avkare Issues Voluntary Nationwide Recall of Sildenafil 100 mg Tablets and Trazodone 100 mg Tablets Due to Product Mix-Up
Date: 12/9/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Avkare has posted a lot recall of Sildenafil 100 mg tablets and Trazodone 100 mg tablets.
About this Recall:
Avkare is voluntarily recalling 1 lot of sildenafil 100 mg tablets and 1 lot of trazodone 100 mg tablets to the consumer level. These products have been recalled due to a product mix-up of the listed 2 separate products inadvertently packaged together during bottling at a third party facility.
What this means to you:
Unintended intake of sildenafil may pose serious health risks to those with underlying medical issues. For example, sildenafil may interact with nitrates found in some prescription drugs (such as nitroglycerin) lowering blood pressure to dangerous levels. Patients with diabetes, high blood pressure, or heart disease often take nitrates.
Unintended intake of trazodone may result in adverse health consequences such as somnolence/sedation, dizziness, constipation, and blurred vision. These adverse events may be more concerning in elderly patients due to an increased risk for falls and driving impairment. To date, Avkare has not received any reports of adverse events related to this recall.
Sildenafil, the active ingredient in Viagra®, which is a phosphodiesterase-5 (PDE-5) inhibitor, is used for the treatment of erectile dysfunction (ED) and is packaged in 100 count bottles (NDC 42291-748-01). Trazodone is indicated for the treatment of major depressive disorder and is packaged in 1,000 count bottles (NDC 42291-834- 10). The recalled lots of sildenafil 100 mg tablet (lot 36884 with an expiration date of 03/2022) and trazodone hydrochloride 100 mg tablet (lot 36783 with an expiration date of 06/2022) were distributed to distributors and wholesalers, and then further distributed nationwide. Avkare has notified its distributors and customers and is arranging for return of all recalled product.
Consumers with questions regarding this recall can contact Avkare at 1-855-361-3993 Monday through Friday (8 am – 4 pm CST). Consumers should contact their physician or healthcare provider (HCP) if they have experienced any problems that may be related to taking or using these drug products.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
This recall is being conducted with the knowledge of the United States (US) Food and Drug Administration (FDA).
For more information regarding this FDA Recall Notification, please refer to the FDA website. FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Avkare Issues Voluntary Nationwide Recall of Sildenafil 100 mg Tablets and Trazodone 100 mg Tablets Due to Product Mix-Up
FDA Publish Date: 12/9/2020
Avkare is voluntarily recalling one (1) lot of sildenafil 100 mg tablets and one (1) lot of trazodone 100 mg tablets to the consumer level. These products have been recalled due to a product mix-up of the listed two separate products inadvertently packaged together during bottling at a third party facility.
| Product |
Lot |
Expiration date |
Bottle count |
NDC Number |
|
Sildenafil Tablets, USP |
36884 |
03/2022 |
100 |
42291-748-01 |
|
Trazodone Tablets, USP |
36783 |
06/2022 |
1,000 |
42291-834-10 |
Sildenafil, the active ingredient in Viagra®, which is a (PDE-5) inhibitor, is used for the treatment of erectile dysfunction (ED) and is packaged in 100 count bottles (NDC 42291-748-01). Trazodone is indicated for the treatment of major depressive disorder (MDD) and is packaged in 1,000 count bottles (NDC 42291-834-10). The impacted lots of sildenafil 100 mg tablet (lot 36884 with an expiration date of 03/2022) and trazodone hydrochloride 100 mg tablet (lot 36783 with an expiration date of 06/2022) were distributed to distributors and wholesalers, and then further distributed nationwide.
Avkare has notified its distributors and customers and is arranging for return of all recalled product of the listed lots. Distributors that have any of the product which is being recalled should contact Customer Service at Avkare at 1-855-361-3993 or email customerservice@avkare.com to arrange for the return of the product.
Consumers with questions regarding this recall can contact Avkare at 1-855-361-3993 Monday through Friday (8 am – 4 pm CST). Consumers should contact their physician or healthcare provider (HCP) if they have experienced any problems that may be related to taking or using these drug products.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the United States (US) Food and Drug Administration (FDA).
Link to FDA recall notification.
Company Contact Information
Consumers: Customer Service at Avkare
- 1-855-361-3993
- customerservice@avkare.com
Media: Clifton Stanfill
- 931-908-2192
Product Photos
- Click here for product photos.
Torrent Issues Voluntary Nationwide Recall of Anagrelide Capsules Due to Dissolution Test Failure
Date: 12/9/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Torrent Pharmaceuticals has posted a lot recall of anagrelide capsules.
About this Recall:
Torrent is voluntarily recalling 1 lot of anagrelide capsules to the consumer level due to dissolution test failure detected during routine quality testing.
What this means to you:
Failed dissolution can result in a slower rate and amount of drug release leading to less anagrelide available in the body. For seriously ill patients with increased platelet counts, less available anagrelide could increase the risk for clotting or bleeding events such as a heart attack or stroke which could be life-threatening. To date, Torrent has not received any reports of adverse events related to this recall.
Anagrelide is used to treat a blood cell disorder called thrombocythemia (also called thrombocytosis), which occurs when your body produces too many platelet cells.
As the risk of harm may be higher if the treatment is stopped immediately without any alternative treatment, patients should contact their pharmacist or physician who can advise them about an alternative treatment before returning their medication.
The product being recalled is listed below and packaged in bottles. It can be identified by checking the product name, manufacturer details, and batch or lot number on the bottle containing the product. NDC
| NDC |
Manufacturer |
Product Description |
Lot/Batch |
Expiration Date |
|
13668-462-01 |
Torrent |
anagrelide capsule, USP 1 mg 100-count bottles |
BFD1G001 |
12/2021 |
Torrent is arranging for return of all recalled products to Qualanex.
Consumers with medical questions regarding this recall or to report an adverse event can contact Torrent at:
- 1-800-912-9561 (live calls received 8:00 am – 5:00 pm Eastern Time, Monday to Friday; voicemail available 8:00 am – 5:00 pm Eastern Time, Monday to Friday).
- Medinfo.Torrent@apcerls.com
Consumers should also contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Any general questions regarding the return of this product should be directed to Qualanex at 1-888-424-4340 (live calls received 8 am – 5 pm Eastern Time, Monday to Friday).
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the United States (US) Food and Drug Administration (FDA).
Torrent Issues Voluntary Nationwide Recall of Anagrelide Capsules, USP Due to Dissolution Test Failure
FDA Publish Date: 12/9/2020
Torrent is voluntarily recalling one (1) lot of anagrelide capsules, USP to the consumer level due to dissolution test failure detected during routine quality testing.
Failed dissolution can result in a slower rate and extent of drug release leading to less anagrelide available in the body. For seriously ill patients with elevated platelet counts, less available anagrelide could increase the risk of clotting or bleeding events such as a heart attack or stroke which could be life-threatening. To date, Torrent has not received any reports of adverse events related to this recall.
Anagrelide is used to treat a blood cell disorder called thrombocythemia (also called thrombocytosis), which occurs when your body produces too many platelets.
As the risk of harm may be higher if the treatment is stopped immediately without any alternative treatment, patients should contact their pharmacist or physician who can advise them about an alternative treatment prior to returning their medication.
The product subjected to the recall is listed below and packaged in bottles. The product can be identified by checking the product name, manufacturer details, and batch or lot number on the bottle containing the product.
| NDC |
Manufacturer |
Product Description |
Lot/Batch |
Expiration Date |
|
13668-462-01 |
Torrent |
anagrelide capsule USP 1 mg, 100-count bottles |
BFD1G001 |
12/2021 |
Anagrelide capsules, USP were distributed nationwide to Torrent’s wholesale distributor and retail customers. Torrent is notifying its distributors and customers by phone and in writing to immediately discontinue distribution of the specific lots being recalled and to notify their sub-accounts. Torrent is arranging for return of all recalled products to Qualanex. Instructions for returning recalled products are given in the recall letter.
Consumers with medical questions regarding this recall or to report an adverse event can contact Torrent at:
• 1-800-912-9561 (live calls received 8:00 am – 5:00 pm Eastern Time, Monday to Friday; voicemail available 8:00 am – 5:00 pm Eastern Time, Monday to Friday).
• Medinfo.Torrent@apcerls.com
2—1209-2020 Class 2 Anagrelide Capsules – Torrent Voluntary Nationwide Recall
Consumers should also contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Any general questions regarding the return of this product should be directed to Qualanex at 1-888-424-4340 (live calls received 8 am – 5 pm Eastern Time, Monday to Friday).
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the United States (US) Food and Drug Administration.
Link to FDA recall notification: https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts/torrent-pharmaceuticals-limited-issues-voluntary-nationwide-recall-anagrelide-capsules-usp-due
Company Contact Information
Consumers: Torrent Pharmaceuticals Limited
- 1-800-912-9561
- Medinfo.Torrent@apcerls.com
Product Photos
Click here for product photos.
Recall of Regenecare® HA Hydrogel
Date: 12/02/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
MPM Medical LLC has posted a lot recall of Regenecare® HA Hydrogel.
About this Recall:
MPM Medical is voluntarily recalling one (1) lot of Regenecare HA Hydrogel to the consumer level. Following two customer complaints of visible contamination, the product was found to be contaminated with the bacteria Burkholderia cepecia.
What this means to you:
Topical application of Regenecare HA Hydrogel contaminated with B. cepacia may result in local skin infections. For immunocompromised patients, including patients receiving chemotherapy and patients with cystic fibrosis, the skin infection is more likely to spread into the blood stream leading to life-threatening sepsis which can include symptoms such as fever, difficulty breathing, low blood pressure, fast heart rate, mental confusion, and possibly death. To date, MPM Medical has not received any reports of adverse events related to this recall.
Regenecare HA Hydrogel is an over-the-counter (OTC) product that contains 2% lidocaine and is used topically for temporary relief of pain and itching associated with minor burns, sunburn, minor cuts, scrapes, insect bites, or minor skin irritations. It is packaged in 3 oz. plastic tubes and distributed in boxes of 12. The product can be identified by NDC 66977-107-03, and the lot number 41262 with expiration date 2021-01 debossed on the tube crimp as shown in the images provided at the hyperlink below. The recalled Regenecare HA Hydrogel lot (#41262) was distributed nationwide to wholesalers and healthcare facilities.
Patients in possession of the product which is being recalled should stop using. Consumers with questions regarding this recall can contact MPM Medical by phone at 1-800-232-5512 (toll-free) Monday through Friday between 7 AM and 5 PM CST. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to using this drug product. For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2
MPM Medical Issues Voluntary Nationwide Recall of Regenecare® HA Hydrogel Due to Burkholderia cepecia Contamination
FDA Publish Date: 12/02/2020
MPM Medical is voluntarily recalling one (1) lot of Regenecare® HA Hydrogel to the consumer level. Following two customer complaints of visible contamination, the product was found to be contaminated with the bacteria Burkholderia cepecia.
Risk Statement: Topical application of Regenecare HA Hydrogel contaminated with B. cepacia may result in local skin infections. For immunocompromised patients, including patients receiving chemotherapy and patients with cystic fibrosis, the skin infection is more likely to spread into the blood stream leading to life-threatening sepsis which can include symptoms such as fever, difficulty breathing, low blood pressure, rapid heart rate, mental confusion, and possibly death. To date, MPM Medical has not received any reports of adverse events related to this recall.
Regenecare HA Hydrogel is an over-the-counter (OTC) product that contains 2% lidocaine and is used topically for temporary relief of pain and itching associated with minor burns, sunburn, minor cuts, scrapes, insect bites, or minor skin irritations. It is packaged in 3 oz. plastic tubes and distributed in boxes of 12. The product can be identified by NDC 66977-107-03 and the lot number 41262 with an expiration date 2021-01 debossed on the tube crimp as shown in the images provided at the below hyperlink. The recalled Regenecare HA Hydrogel lot (#41262) was distributed nationwide to wholesalers and healthcare facilities.
MPM Medical is notifying its distributors and customers by first class mail, electronic mail, and phone call and is arranging for return of all recalled product. Patients and healthcare facilities in possession of this product which is being recalled should stop using and dispensing.
Consumers with questions regarding this recall can contact MPM Medical by phone at 1-800-232-5512 (toll-free) Monday through Friday between 7 AM and 5 PM CST. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
MPM Medical is committed to delivering safe, fully compliant products of the highest quality and is taking necessary steps to prevent future occurrence of this issue.
This recall is being conducted with the knowledge of the United States (US) Food and Drug Administration.
Company Contact Information
Consumers:
- MPM Medical, LLC
- 1-800-232-5512
Product Photos
- To view product photos, use link to FDA website
Nostrum Issues Voluntary Nationwide Recall of Metformin HCl Extended Release Tablets, USP 500 mg and 750 mg, Due to NDMA Content Above the Acceptable Daily Intake Limit
Date: 11/2/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Nostrum Laboratories has posted a lot recall of metformin.
About this Recall:
Nostrum Laboratories is recalling 2 lots of Metformin Hydrochloride (HCl) Extended Release (ER) Tablets, USP 500 mg and 2 lots of Metformin HCl ER Tablets, USP 750 mg. These products are being recalled as they contain levels of nitrosamine impurities above the Acceptable Daily Intake (ADI) limit of 96 ng/day.
What this means to you:
NDMA is classified as a probable human carcinogen (a substance that could cause cancer) based on results from laboratory tests. NDMA is a known environmental contaminant and is found in water and foods (including meats, dairy products, and vegetables). Nostrum has not received any reports of adverse events related to this recall.
Metformin HCl ER tablets are indicated as an adjunct to diet and exercise to improve blood glucose control in adults with type 2 diabetes mellitus. Product is packaged in HDPE bottles of 100 tablets, with NDC 29033-055-01 (500 mg) or NDC 29033-056-01 (750 mg). The 500 mg product can be identified as an off-white oblong tablet debossed with “NM5”, and the 750 mg product can be identified as an off-white oblong tablet debossed with “NM7”. The impacted lots of Metformin HCl ER Tablets, USP 500 mg and 750 mg can be found at the FDA Recall Notification links on the following page.
Consumers should consult a healthcare professional (HCP) to obtain a replacement or a different treatment option. It could be dangerous for patients with type 2 diabetes to stop taking their metformin without first talking to their HCP. Consumers should contact their physician or HCP if they have experienced any problems that may be related to taking this drug product.
Consumers with medical questions regarding this recall can contact Nostrum Medical Affairs at phone number 816-308-4941 or via email at quality@nostrumpharma.com Monday through Friday from 8 am to 5 pm CST. Consumers should contact their physician or pharmacy for further medical advice.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online.
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
This recall is being conducted with the knowledge of the United States (US) Food and Drug Administration.
Nostrum Issues Voluntary Nationwide Recall of Metformin HCl Extended Release Tablets, USP 500 mg and 750 mg, Due to NDMA Content Above the Acceptable Daily Intake Limit
Date: 11/2/2020
Nostrum Laboratories, Inc. is voluntarily recalling 2 lots of Metformin Hydrochloride (HCl) Extended Release (ER) Tablets, USP 500 mg and 2 lots of Metformin HCl ER Tablets, USP 750 mg to the consumer level. The Metformin HCl ER Tablets, USP 500 mg and 750 mg have been found to contain levels of nitrosamine impurities above the Acceptable Daily Intake (ADI) limit of 96 ng/day as published in the FDA Guidance Document issued September 2020.
NDMA is classified as a probable human carcinogen (a substance that could cause cancer) based on results from laboratory tests. NDMA is a known environmental contaminant and is found in water and foods (including meats, dairy products, and vegetables). Nostrum has not received any reports of adverse events related to this recall.
Metformin HCl ER tablets are indicated as an adjunct to diet and exercise to improve blood glucose control in adults with type 2 diabetes mellitus. Product is packaged in HDPE bottles of 100 tablets, with NDC 29033-055-01 (500 mg) or NDC 29033-056-01 (750 mg). The affected Metformin HCl ER Tablets, USP 500 mg and 750 mg lots are listed in the table below.
| Product Description |
NDC |
Lot Numbers |
Expiry Dates |
|
Metformin HCl Extended |
29033-055-01 |
MET100201 |
05/2022 |
|
MET100401 |
05/2022 |
||
|
Metformin HCl Extended |
29033-056-01 |
MET200101 |
05/2022 |
|
MET200301 |
05/2022 |
Nostrum is notifying its distributors by letter and is arranging for return of all recalled products. Pharmacies that have Metformin HCl ER Tablets, USP 500 mg or 750 mg which are being recalled should return to the place of purchase. Consumers should consult a healthcare professional (HCP) to obtain a replacement or a different treatment option. It could be dangerous for patients with type 2 diabetes to stop taking their metformin without first talking to their HCP. Consumers should contact their physician or HCP if they have experienced any problems that may be related to taking this drug product.
Consumers with medical questions regarding this recall can contact Nostrum Medical Affairs at phone number 816-308-4941 or via email at quality@nostrumpharma.com Monday through Friday from 8 am to 5 pm CST. Consumers should contact their physician or pharmacy for further medical advice.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online.
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
This recall is being conducted with the knowledge of the United States (US) Food and Drug Administration.
Links to FDA recall notifications
Company Contact Information
Consumers: Nostrum Laboratories, Inc. Medical Affairs
- 816-308-4941
- quality@nostrumpharma.com
Sunstar Americas Issues Voluntary Nationwide Recall of Paroex® Chlorhexidine Gluconate Oral Rinse USP, 0.12% Due to Microbial Contamination
Date: 10/28/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Sunstar Americas, Inc. (SAI) has posted a lot recall of Paroex.
About this Recall:
SAI is voluntarily recalling specific lots of Paroex® Chlorhexidine Gluconate Oral Rinse USP, 0.12% with an expiration date from 6/30/2022 to 9/30/2022 to the consumer level. This product is being recalled as it may be contaminated with the bacteria Burkholderia lata.
What this means to you:
Use of the contaminated product in a person with a normal immune response may result in oral and, potentially, systemic infections requiring antibacterial therapy. In the most at-risk populations, the use of the contaminated product may result in life-threatening infections, such as pneumonia and bacteremia. To date, no adverse events have been reported related to this recall.
The prescription oral rinse product, available through healthcare providers (HCPs) only, is indicated for use as part of a professional program for the treatment of gingivitis and is packaged as follows:
- 1789P GUM® Paroex® is distributed in cases each containing 6 amber bottles of 16 fluid ounce (473 mL) chlorhexidine rinse. The bottle has a childproof cap and a 15 mL metered dosage cup, is safety sealed, and is decorated with a multiple-panel wrap-around label.
- 1788P GUM® Paroex® is distributed in cases each containing 24 amber bottles of 4 fluid ounce (118.25 mL) chlorhexidine rinse. The bottle has a childproof cap, is safety sealed, and is decorated with a multiple-panel wrap-around label.
The product can be identified as shown in the images linked here.
Paroex was distributed nationwide to dental offices, dental distributors, pharmaceutical wholesalers, dental schools, and pharmacies.
SAI is notifying its direct distributors and customers by USPS Priority mail and is arranging for return of all recalled products. Patients, pharmacies, and healthcare facilities in possession of these products should stop using and dispensing immediately.
Consumers with questions regarding this recall can contact SAI by phone at 1-800-528-8537 or email at us.pcr@us.sunstar.com Monday through Friday from 8 am to 5 pm CST. Consumers should contact their physician or HCP if they have experienced any problems that may be related to using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
Sunstar is committed to delivering safe, fully compliant products of the highest quality and is taking necessary steps to prevent future occurrence of this issue.
This recall is being conducted with the knowledge of the US Food and Drug Administration (FDA).
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Sunstar Americas Issues Voluntary Nationwide Recall of Paroex® Chlorhexidine Gluconate Oral Rinse USP, 0.12% Due to Microbial Contamination
FDA Publish Date: 10/28/2020
Sunstar Americas, Inc. (SAI) is voluntarily recalling specific lots of Paroex® Chlorhexidine Gluconate Oral Rinse USP, 0.12% with an expiration date from 6/30/2022 to 9/30/2022 to the consumer level. This product is being recalled as it may be contaminated with the bacteria Burkholderia lata.
Use of the defective product in an immunocompetent person may result in oral and, potentially, systemic infections requiring antibacterial therapy. In the most at-risk populations, the use of the defective product may result in life-threatening infections, such as pneumonia and bacteremia. To date, no adverse events have been reported related to this recall.
The prescription oral rinse product, available through healthcare providers (HCPs) only, is indicated for use as part of a professional program for the treatment of gingivitis and is packaged as follows:
- 1789P GUM® Paroex® is distributed in cases each containing 6 amber bottles of 16 fluid ounce (473 mL) chlorhexidine rinse. The bottle has a childproof cap and a 15 mL metered dosage cup, is safety sealed, and is decorated with a multiple-panel wrap-around label.
- 1788P GUM® Paroex® is distributed in cases each containing 24 amber bottles of 4 fluid ounce (118.25 mL) chlorhexidine rinse. The bottle has a childproof cap, is safety sealed, and is decorated with a multiple-panel wrap-around label.
The product can be identified as shown in the images linked at the bottom of this memo.
Paroex was distributed nationwide to dental offices, dental distributors, pharmaceutical wholesalers, dental schools, and pharmacies.
SAI is notifying its direct distributors and customers by USPS Priority mail and is arranging for return of all recalled products. Patients, pharmacies, and healthcare facilities in possession of these products should stop using and dispensing immediately.
Consumers with questions regarding this recall can contact SAI by phone at 1-800-528-8537 or email at us.pcr@us.sunstar.com Monday through Friday from 8 am to 5 pm CST. Consumers should contact their physician or HCP if they have experienced any problems that may be related to using this drug product.
Affected products and lot numbers are listed below:
|
Product name: Paroex® Chlorhexidine Gluconate Oral Rinse USP, 0.12% NDC 052376-021-02 P/N 1789P 16 fl. oz. |
|||||
|
Lot # |
Expiration Date |
Lot # |
Expiration Date |
Lot # |
Expiration Date |
|
C170FY |
6/30/22 |
C191KT |
7/31/22 |
C205BL |
7/31/22 |
|
C170FZ |
6/30/22 |
C191KU |
7/31/22 |
C205BM |
7/31/22 |
|
C170GA |
6/30/22 |
C191KW |
7/31/22 |
C205BN |
7/31/22 |
|
C170GB |
6/30/22 |
C191KX |
7/31/22 |
C219DS |
8/31/22 |
|
C170GC |
6/30/22 |
C191KY |
7/31/22 |
C240GM |
9/30/22 |
|
C177GP |
6/30/22 |
C198LJ |
7/31/22 |
C219DK |
8/31/22 |
|
C177GQ |
6/30/22 |
C198LK |
7/31/22 |
C219DL |
8/31/22 |
|
C177GR |
6/30/22 |
C198LL |
7/31/22 |
C219DM |
8/31/22 |
|
C240GP |
9/30/22 |
C198LM |
7/31/22 |
C219DN |
8/31/22 |
|
C240GQ |
9/30/22 |
C205BH |
7/31/22 |
C219DP |
8/31/22 |
|
C240GR |
9/30/22 |
C205BJ |
7/31/22 |
C219DQ |
8/31/22 |
|
C191KS |
7/31/22 |
C205BK |
7/31/22 |
C219DR |
8/31/22 |
|
Product name: Paroex® Chlorhexidine Gluconate Oral Rinse USP, 0.12% NDC 052376-021-04 P/N 1788P 4 fl. oz. |
|||||
|
Lot # |
Expiration Date |
||||
|
C191KR |
7/31/22 |
3—1028-2020 Class 2 Paroex – Sunstar Voluntary Nationwide Recall
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
• Complete and submit the report Online
• Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
Sunstar is committed to delivering safe, fully compliant products of the highest quality and is taking necessary steps to prevent future occurrence of this issue.
This recall is being conducted with the knowledge of the US Food and Drug Administration (FDA).
About Sunstar Americas Inc.
Sunstar Americas, Inc., a member of the Sunstar Group of companies, is a global organization headquartered in Switzerland that is a leader in the oral care industry and the manufacturer and distributor of the GUM and Butler Brands.
For more information:
us.pcr@us.sunstar.com Contact Sunstar: Phone: 1-800-528-8537 Email:
https://www.gumbrand.com/news-announcements Website:
Link to FDA recall notification: https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts/sunstar-americas-inc-issues-voluntary-nationwide-recall-paroexr-chlorhexidine-gluconate-oral-rinse
Company Contact Information
Consumers: Sunstar Americas, Inc.
- 1-800-528-8537
- us.pcr@us.sunstar.com
Product Photos
Marksans Pharma Issues Expansion of Voluntary Nationwide Recall of Metformin HCl ER Tablets, USP 500 mg & 750 mg, Due to the Detection of NDMA
Date: 10/5/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Marksans Pharma has posted a lot recall of metformin.
About this Recall:
Marksans Pharma is voluntarily expanding its earlier initiated recall on June 5, 2020 to include an additional 76 unexpired lots of Metformin Hydrochloride (HCl) Extended-Release (ER) Tablets, USP 500 mg and 750 mg to the consumer level. Marksans performed N-Nitrosodimethylamine (NDMA) testing of unexpired identified marketed lots and observed that NDMA content in some lots was exceeding the acceptable daily intake (ADI) limit of 96 ng/day, therefore, out of an abundance of caution, an additional 76 lots are being recalled.
What this means to you:
NDMA is classified as a substance that could cause cancer based on results from laboratory tests. NDMA is a known environmental contaminant found in water and foods (such as meats, dairy products, and vegetables). Marksans has not received any reports of adverse events that have been related to this recall.
Metformin HCl ER Tablets, USP 500 mg and 750 mg are indicated as an adjunct to diet and exercise to improve blood glucose control in adults with type 2 diabetes mellitus. It is packaged in bottles with the following NDC numbers in different packing configurations.
Metformin HCl ER Tablets, USP 500 mg:
- 90 counts: 49483-0623-09
- 100 counts: 49483-0623-01
- 500 counts: 49483-0623-50
- 1,000 counts: 49483-0623-10
Metformin HCl ER Tablets, USP 750 mg:
- 100 counts: 49483-0624-01
The affected Metformin HCl ER Tablets, USP 500 mg, are white to off white, capsule shaped, biconvex tablets, debossed with ‘101’ on one side and plain on the other side. The impacted Metformin HCl ER Tablets, USP 750 mg, are white to off white, capsule shaped, biconvex tablets, debossed with ‘102’ on one side and plain on the other side.
For a complete listing of recalled lot numbers and expiration dates, view the FDA recall notification: https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts/marksans-pharma-limited-issues-expansion-voluntary-nationwide-recall-metformin-hydrochloride.
Consumers taking these recalled product lots of metformin ER tablets are instructed by the FDA to continue taking it, until a doctor or pharmacist gives them a replacement or an alternative treatment option. It could be dangerous for patients with type 2 diabetes to stop taking their metformin without first talking to their healthcare professional (HCP). Consumers should contact their physician or HCP if they have experienced any problems that may be related to taking or using this drug product.
Consumers with questions regarding this recall and return can contact Ms. Irene McGregor (Vice President, Regulatory Affairs) of Time-Cap Labs by phone number 631-753-9090, ext. 160, Monday to Friday 8 am to 5 pm EST or by e-mail at imcgregor@timecaplabs.com.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
Link to initial FDA recall notification
Link to expanded FDA recall notification
For more information regarding this FDA Recall Notification, please refer to the FDA website
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
2020 Magellan Rx Management, LLC. All rights reserved.
Marksans Issues Expansion of Voluntary Nationwide Recall of Metformin HCl ER Tablets, USP 500 mg & 750 mg, Due to the Detection of NDMA
FDA Publish Date: 10/5/2020
Marksans Pharma is voluntarily expanding its earlier initiated recall on June 5, 2020 to include an additional 76 unexpired lots of Metformin Hydrochloride (HCl) Extended-Release (ER) Tablets, USP 500 mg and 750 mg to the consumer level. Marksans performed N-Nitrosodimethylamine (NDMA) testing of unexpired identified marketed lots and observed that the NDMA content in some lots was exceeding the acceptable daily intake limit (ADI) of 96 ng/day, therefore, out of an abundance of caution, an additional 76 lots are being recalled.
Risk Statement: NDMA is classified as a probable human carcinogen (a substance that could cause cancer) based on results from laboratory tests. NDMA is a known environmental contaminant found in water and foods (such as meats, dairy products, and vegetables). Marksans has not received any reports of adverse events related to this recall.
Metformin HCl ER Tablets, USP 500 mg and 750 mg are indicated as an adjunct to diet and exercise to improve blood glucose control in adults with type 2 diabetes mellitus. It is packaged in HDPE bottles with the following NDCs in different packing configurations:
Metformin HCl ER Tablets, USP 500 mg:
- 90 counts: 49483-0623-09
- 100 counts: 49483-0623-01
- 500 counts: 49483-0623-50
- 1,000 counts: 49483-0623-10
Metformin HCl ER Tablets, USP 750 mg:
- 100 counts: 49483-0624-01
The affected Metformin HCl ER Tablets, USP 500 mg, are white to off white, capsule shaped, biconvex tablets, debossed with ‘101’ on one side and plain on the other side. The impacted Metformin HCl ER Tablets, USP 750 mg, are white to off white, capsule shaped, biconvex tablets, debossed with ‘102’ on one side and plain on the other side.
For a complete listing of recalled lot numbers and expiration dates, view the FDA recall notification: https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts/marksans-pharma-limited-issues-expansion-voluntary-nationwide-recall-metformin-hydrochloride.
Metformin HCl ER Tablets, USP 500 mg and 750 mg were distributed by Time-Cap Labs nationwide in the United States (US) to wholesalers who further distributed to pharmacies. Marksans is notifying its distributors and customers by issuing a notification letter and press release and is arranging for return/replacement of recalled product lots. Distributors and customers that have affected lots of metformin HCl ER tablets that are being recalled should return to the place of purchase. The lot number can be located on the side panel of bottle labels as well as shipper/case labels.
Consumers taking these recalled product lots of metformin ER tablets are instructed by the FDA to continue taking it, until a doctor or pharmacist gives them a replacement or an alternative treatment option. It could be dangerous for patients with type 2 diabetes to stop taking their metformin without first talking to their healthcare professional (HCP). Consumers should contact their physician or HCP if they have experienced any problems that may be related to taking or using this drug product. Consumers with questions regarding this recall and return can contact Ms. Irene McGregor (Vice President, Regulatory Affairs) of Time-Cap Labs by phone number 631-753-9090, ext. 160, Monday to Friday 8 AM to 5 PM EST or by e-mail at imcgregor@timecaplabs.com.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
• Complete and submit the report Online
• Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the US Food and Drug Administration (FDA).
Company Contact Information
Consumers: Ms. Irene McGregor (Vice President, Regulatory Affairs) of Time-Cap Labs, Inc.
- 631-753-9090; ext. 160
- imcgregor@timecaplabs.com
Product Photos
Sun Pharma Issues Voluntary Nationwide Recall of Riomet ER™ (metformin HCl for ER oral suspension) due to NDMA
Date: 9/23/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Sun Pharma has posted a lot recall of Riomet ER™ (metformin hydrochloride [HCl] for extended-release [ER] oral suspension).
About this Recall:
Sun Pharma is voluntarily recalling 1 lot of Riomet ER (metformin HCl for ER oral suspension), 500 mg per 5 mL, to the consumer level. The reason for the recall is due to the level of N-Nitrosodimethylamine (NDMA), which has been found to be above the acceptable daily intake (ADI) limit established by the United States (US) Food and Drug Administration (FDA).
NDMA is classified as a substance that could cause cancer based on results from laboratory tests. NDMA is a known environmental contaminant and is found in water and foods, such as meats, dairy products, and vegetables. To date, Sun Pharma has not received any reports of adverse events related to this recall.
What this means to you:
Patients taking Riomet ER (metformin HCl for ER oral suspension), 500 mg per 5 mL are advised to continue taking their medication and contact their pharmacist, physician, or medical provider for advice regarding an alternative treatment. According to the US FDA, it could be dangerous for patients with this serious condition to stop taking their metformin without first talking to their healthcare professionals (HCPs). Please visit the agency’s website for more information at https://www.fda.gov/drugs/drug-safety-andavailability/fda-updates-and-press-announcements-ndma-metformin.
Riomet ER (metformin HCl for ER oral suspension) is a prescription oral medication indicated as an add-on to diet and exercise to improve blood glucose control in adults and pediatric patients 10 years of age and older with type 2 diabetes mellitus. Riomet ER, when reconstituted, is packaged in a 16 oz. (473 mL) round bottle. Each carton contains 1 bottle of drug pellets, 1 bottle of diluent, and 1 dosing cup.
The product can be identified by the bottles or carton labeled as Riomet ER containing the specific NDC, lot number, and expiration date found at the FDA Recall Notification link below.
Consumers with questions regarding this recall can contact Sun Pharma by calling 1-800-818-4555 Monday through Friday between 8:00 am to 5:00 pm EST or e-mailing drug.safetyUSA@sunpharma.com. Consumers should contact their physician or HCP if they have experienced any problems that may be related to taking or using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Sun Pharma Issues Voluntary Nationwide Recall of Riomet ER™ (metformin HCl for ER oral suspension) due to NDMA
FDA Publish Date: 9/23/2020
Sun Pharma is voluntarily recalling 1 lot of Riomet ER™ (metformin hydrochloride [HCl] for extended-release [ER] oral suspension), 500 mg per 5 mL, to the consumer level. The reason for the recall is due to the level of N-Nitrosodimethylamine (NDMA), which has been found to be above the acceptable daily intake (ADI) limit established by the United States (US) Food and Drug Administration (FDA).
NDMA is classified as a probable human carcinogen (a substance that could cause cancer) based on results from laboratory tests. NDMA is a known environmental contaminant and is found in water and foods, such as meats, dairy products, and vegetables. To date, Sun Pharma has not received any reports of adverse events related to this recall.
Riomet ER (metformin HCl for ER oral suspension) is a prescription oral medication indicated as an adjunct to diet and exercise to improve glycemic control in adults and pediatric patients 10 years of age and older with type 2 diabetes mellitus. Riomet ER, when reconstituted, is packaged in a 16 oz. (473 mL) round bottle. Each carton contains 1 bottle of drug pellets, 1 bottle of diluent, and 1 dosing cup. The affected Riomet ER is the following lot:
|
Product Name |
Lot Number |
NDC Number |
Expiration Date |
Number of Units |
|
Riomet ER (metformin HCl for ER oral suspension), 500 mg per 5 mL |
AB06381 |
10631-019-17 |
10/2021 |
747 cartons |
The product can be identified by the bottles or carton labeled as Riomet ER (metformin HCl for ER oral suspension), containing the specific lot number and expiration date referenced above or on the product photos linked below. The product was distributed nationwide to wholesale customers.
Sun Pharma is notifying its distributors and customers through its third-party Recall Coordinator (Inmar), via FedEx standard overnight shipping and will arrange for return of all recalled products.
Distributors and retailers that have Riomet ER (metformin HCl for ER oral suspension), which is being recalled, should stop distributing and return it to the place of purchase or as directed in the recall notification.
Patients taking Riomet ER (metformin HCl for ER oral suspension), 500 mg per 5 mL are advised to continue taking their medication and contact their pharmacist, physician, or medical provider for advice regarding an alternative treatment. According to the US FDA, it could be dangerous for patients with this serious condition to stop taking their metformin without first talking to their healthcare professionals (HCPs). Please visit the agency’s website for more information at https://www.fda.gov/drugs/drug-safety-andavailability/fda-updates-and-press-announcements-ndma-metformin.
Consumers with questions regarding this recall can contact Sun Pharma by calling 1-800-818-4555 Monday through Friday between 8:00 am to 5:00 pm EST or e-mailing drug.safetyUSA@sunpharma.com. Consumers should contact their physician or HCP if they have experienced any problems that may be related to taking or using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
This recall is being conducted with the knowledge of the US FDA.
Link to FDA recall notification
Company Contact Information
Consumers: Sun Pharma
- 1-800-818-4555
- Drug.safetyUSA@sunpharma.com
Media: Vinita Alexander
- 1-609-720-8197
- vinita.alexander@sunpharma.com
Product Photos
FDA Alerts of Perrigo’s Voluntary Albuterol Inhaler Recall
Date: 9/21/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Perrigo has posted a market recall of albuterol inhalers.
About this Recall:
The United States Food and Drug Administration (FDA) is alerting healthcare professionals (HCPs) and patients of a voluntary recall of all unexpired albuterol sulfate inhalation aerosol manufactured by Catalent Pharma for Perrigo due to possible clogging of the inhaler resulting in patients not receiving enough or any medicine.
What this means to you:
The albuterol inhaler delivers medication into the body through the airway and lungs, where it opens the airways to treat asthma and other conditions, such as chronic obstructive pulmonary disease (COPD). Patients could experience health risks if their rescue albuterol inhaler malfunctions and does not relieve symptoms in an emergency situation. The FDA urges patients to continue using the inhaler they have on hand.
Additionally, the FDA advises patients to:
- immediately seek emergency care if needed;
- use their Perrigo inhaler they have on hand, as needed and as directed by a doctor;
- have extra inhalers or an alternative treatment available in case of malfunction, as some of these recalled inhalers stop working after several uses; and
- contact their HCP or pharmacist with questions.
FDA reminds HCPs and patients that albuterol inhalers are available through additional manufacturers. Perrigo informed the FDA it had received several thousand complaints about its product. Most of the complaints were for clogging and failure to dispense enough medicine. The manufacturer of Perrigo’s albuterol inhaler, Catalent, stopped producing and distributing the albuterol inhaler products on August 21, 2020 and is currently investigating the malfunction.
The agency asks HCPs and patients to report unexpected side effects or quality problems associated with albuterol inhalers to FDA’s MedWatch Adverse Event Reporting program:
- Complete and submit the report online at www.fda.gov/medwatch/report.htm; or
- Download and complete the form, then submit it via fax at 1-800-FDA-0178.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
FDA Alerts of Perrigo’s Voluntary Albuterol Inhaler Recall
FDA Publish Date: 9/21/2020
The United States (US) Food and Drug Administration (FDA) is alerting healthcare professionals (HCPs) and patients of a voluntary recall of all unexpired albuterol sulfate inhalation aerosol manufactured by Catalent Pharma for Perrigo, due to possible clogging of the inhaler resulting in patients not receiving enough or any medicine. This recall is to the retail level. The FDA urges patients to continue using the inhaler they have on hand.
The albuterol inhaler delivers medication into the body through the airway and lungs, where it opens the airways to treat asthma and other conditions, such as chronic obstructive pulmonary disease (COPD). Patients could experience health risks if their rescue albuterol inhaler malfunctions and does not relieve symptoms in an emergency situation.
The FDA is advising patients to:
- immediately seek emergency care if needed;
- use their Perrigo inhaler they have on hand, as needed and as directed by a doctor;
- have extra inhalers or an alternative treatment available in case of malfunction, as some of these recalled inhalers stop working after several uses; and
- contact their HCP or pharmacist with questions.
FDA reminds HCPs and patients that albuterol inhalers are available through additional manufacturers.
Perrigo informed the FDA it had received several thousand complaints about its product. Most of the complaints were for clogging and failure to dispense enough medicine. The manufacturer of Perrigo’s albuterol inhaler, Catalent, stopped producing and distributing the albuterol inhaler products on August 21, 2020 and is currently investigating the malfunction.
The agency asks HCPs and patients to report unexpected side effects or quality problems associated with albuterol inhalers to FDA’s MedWatch Adverse Event Reporting program:
- Complete and submit the report online at www.fda.gov/medwatch/report.htm; or
- Download and complete the form, then submit it via fax at 1-800-FDA-0178.
Link to FDA recall notification
RLC Labs Issues Voluntary Nationwide Recall of All Lots of Nature-Throid® and WP Thyroid® Due to Sub Potency
Date: 9/3/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
RLC Labs has posted a recall of all lots of Nature-Throid and WP Thyroid.
About this Recall:
RLC Labs is voluntarily recalling a total of 483 lots of Nature-Throid and WP Thyroid in all strengths and all counts that are currently within expiration. The products are being recalled as testing of samples from 6 lots by the United States (US) Food and Drug Administration (FDA) found the samples to contain lower levels of drug than expected or to be sub potent. The product may have as low as 87% of the labeled amount of thyroid hormones, liothyronine (T3) or levothyroxine (T4).
What this means to you:
Patients being treated for hypothyroidism (underactive thyroid), who receive recalled Nature-Throid or WP Thyroid, may experience signs and symptoms of underactive thyroid which may include fatigue, increased sensitivity to cold, constipation, dry skin, puffy face, hair loss, slow heart rate, depression, swelling of the thyroid gland, and/or unexplained weight gain or difficulty losing weight. There is reasonable risk of serious injury in newborn infants or pregnant women with hypothyroidism. In elderly patients and patients with underlying cardiac disease, toxic cardiac manifestations of abnormal thyroid levels may occur. RLC Labs has not received any reports of adverse events related to this recall.
Nature-Throid® and WP Thyroid® (thyroid tablets, USP) are composed of liothyronine and levothyroxine and are used to treat underactive thyroid. The manufacturer is instructing patients who are currently taking Nature-Throid and WP Thyroid not to discontinue use without contacting their healthcare provider for further guidance and/or a replacement prescription.
Consumers with questions about the recall can email RLC Labs at recall@rlclabs.com or contact RLC Labs’ Customer Service at 1-877-797-7997, Monday through Thursday from 7:00 AM to 4:00 PM MST and Friday from 7:00 AM to 3:00 PM MST. To identify recalled product, the NDCs, product descriptions, lot numbers, and expiration dates are listed in the document found .
Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
RLC Labs Issues Voluntary Nationwide Recall of All Lots of Nature-Throid® and WP Thyroid® Due to Sub Potency
FDA Publish Date: 9/3/2020
RLC Labs is voluntarily recalling a total of 483 lots of Nature-Throid and WP Thyroid in all strengths and all counts that are currently within expiry to the consumer level. The products are being recalled as testing of samples from 6 lots by the United States (US) Food and Drug Administration (FDA) found the samples to be sub potent. The product may have as low as 87% of the labeled amount of liothyronine (T3) or levothyroxine (T4).
Risk Statement: Patients being treated for hypothyroidism (underactive thyroid), who receive sub potent Nature-Throid or WP Thyroid, may experience signs and symptoms of underactive thyroid which may include fatigue, increased sensitivity to cold, constipation, dry skin, puffy face, hair loss, slow heart rate, depression, swelling of the thyroid gland, and/or unexplained weight gain or difficulty losing weight. There is reasonable risk of serious injury in newborn infants or pregnant women with hypothyroidism. In elderly patients and patients with underlying cardiac disease, toxic cardiac manifestations of abnormal thyroid levels may occur. RLC Labs has not received any reports of adverse events related to this recall.
Nature-Throid® and WP Thyroid® (thyroid tablets, USP) are composed of liothyronine and levothyroxine and are used to treat hypothyroidism. The products subject to recall are packaged in 30, 60, 90, 100, and 1,000 count bottles.
To identify recalled product, the NDCs, product descriptions, lot numbers, and expiration dates are listed in the document found here. These lots were distributed nationwide in the US to RLC Labs’ direct accounts, including healthcare professionals and retail pharmacies.
RLC Labs is proactively notifying its wholesalers by email, mail, and phone to discontinue distribution of the product being recalled and is arranging for the return of all recalled products. The manufacturer is instructing patients who are currently taking Nature-Throid and WP Thyroid not to discontinue use without contacting their healthcare provider for further guidance and/or a replacement prescription.
Consumers with questions about the recall can email RLC Labs at recall@rlclabs.com or contact RLC Labs’ Customer Service at 1-877-797-7997, Monday through Thursday from 7:00 AM to 4:00 PM MST and Friday from 7:00 am to 3:00 pm MST.
Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the US Food and Drug Administration.
Link to FDA recall notification
Company Contact Information
Consumers: RLC Labs, Inc.
- recall@rlclabs.com
Product Photos
Metformin ER 500 mg and 750 mg Tablets
Date: 08/20/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Bayshore has posted a lot recall of Metformin Hydrochloride (HCl) Extended-Release (ER) Tablets, 500 mg and 750 mg.
About this Recall:
Bayshore Pharmaceuticals is voluntarily recalling one (1) lot of metformin HCl ER tablets, 500 mg, and one (1) lot of metformin HCl ER tablets, 750 mg, within expiration to the consumer level. The recall is due to N-nitroso-dimethylamine (NDMA) levels above the acceptable daily intake (ADI) limit. This product was manufactured by Beximco in June 2019, for United States (US) distribution by Bayshore.
What this means to you:
NDMA is a substance that could cause cancer based on results from laboratory tests. NDMA is a known environmental contaminant and is found in water and foods (such as meats, dairy products, and vegetables). The impacted metformin ER tablets can be identified by the NDC number found on the product label and the product lot number. The recalled NDC and lot numbers are listed on the FDA Recall Notification at the link below. Patients who have received impacted lots of metformin ER tablets are advised by the manufacturer to continue taking their medication and contact their pharmacist, physician, or medical provider for advice regarding an alternative treatment. According to the FDA, it could be dangerous for patients with this serious condition to stop taking their metformin without first talking to their healthcare professionals.
Patients with medical-related questions, who wish to report an adverse event or quality issue about the products being recalled should contact Bayshore by phone at 877-372-6093.
Patients wishing to return product may contact Bayshore’s product recall processor, Qualanex, to obtain instructions and a return kit for returning their medication:
- Contact Qualanex at 888-504-2013
- Qualanex will provide the materials needed to return their medication and instructions for reimbursement
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Bayshore Pharmaceuticals Issues Voluntary Nationwide Recall of Metformin ER Tablets 500 mg and 750 mg Due to NDMA Impurity
FDA Publish Date: 08/20/2020
Bayshore Pharmaceuticals is voluntarily recalling one (1) lot of Metformin Hydrochloride (HCl) Extended-Release (ER) Tablets USP, 500 mg, in 1000-count bottles and one (1) lot of Metformin HCl ER Tablets USP, 750 mg, in 100-count bottles within expiry to the consumer level due to the detection of N-Nitrosodimethylamine (NDMA) levels above the acceptable daily intake (ADI) Limit. This product was manufactured by Beximco in June 2019, for United States (US) distribution by Bayshore.
Bayshore was notified by the US Food and Drug Administration (FDA) that 1 lot (lot #18657) of metformin HCl ER tablets, 750 mg, was tested and showed NDMA levels in excess of the ADI limit and recommended recall of the 1 tested lot.
Bayshore has agreed to recall this lot, and out of an abundance of caution, the company has tested samples from 8 lots of metformin HCl ER tablets manufactured using same active pharmaceutical ingredient (API) lot of the failed lot. Out of 8 lots, 1 lot (lot #18657) of metformin HCl ER tablets, 750 mg, and 1 lot (lot #18641) of metformin HCl ER tablets, 500 mg, have showed NDMA levels in excess of the ADI limit. As a result, Bayshore has decided to recall the 2 lots. To date, neither Bayshore nor Beximco have received any reports of adverse events related to use of the product.
Risk Statement: NDMA is classified as a probable human carcinogen (a substance that could cause cancer) based on results from laboratory tests. NDMA is a known environmental contaminant and is found in water and foods (such as meats, dairy products, and vegetables).
Metformin HCl ER tablets USP, 500 mg and 750 mg, are indicated as an adjunct to diet and exercise to improve blood sugar control in adults with type 2 diabetes mellitus. Patients who have received impacted lots of metformin ER tablets are advised by the manufacturer to continue taking their medication and contact their pharmacist, physician, or medical provider for advice regarding an alternative treatment. According to the FDA, it could be dangerous for patients with this serious condition to stop taking their metformin without first talking to their healthcare professionals. Please visit the agency’s website for more information
The metformin HCl ER tablets, 500 mg and 750 mg, lots subject to the recall are identified in the table below.
|
Product Name |
Strength |
Bottle Size |
NDC Number |
Lot # |
Expiration |
|
Metformin HCl ER Tablets USP, 500 mg |
500 mg |
1000-count |
76385-0128-10 |
18641 |
May 2021 |
|
Metformin HCl ER Tablets USP, 750 mg |
750 mg |
100-count |
76385-0129-01 |
18657 |
May 2021 |
The impacted metformin HCl ER tablet lots were distributed nationwide in the US by Bayshore directly to wholesalers and distributors. Bayshore is in the process of notifying its customers impacted by this recall by phone and through recall notification and is arranging for return of the recalled product. Anyone with an existing inventory of the product should quarantine the recalled lots immediately.
Customers and patients with medical-related questions, who wish to report an adverse event or quality issue about the products being recalled should contact Bayshore by phone at 877-372-6093.
Patients wishing to return product may contact Bayshore’s product recall processor, Qualanex, to obtain instructions and a return kit for returning their medication:
- Contact Qualanex at 888-504-2013
- Qualanex will provide the materials needed to return their medication and instructions for reimbursement
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
Patient safety and product quality are critical to Bayshore. Bayshore will continue to partner with, and regularly update, all relevant regulatory authorities as relevant information becomes available.
Company Contact Information
Consumers: Qualanex, Bayshore Pharmaceuticals LLC Information
- 888-504-2013
- 877-372-6093
Product Photos
Recall of Desmopressin Nasal Sprays by Ferring Pharmaceuticals
Date: 08/05/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Ferring Pharmaceuticals has posted a market recall of DDAVP® Nasal Spray 10 mcg/0.1 mL, Desmopressin Acetate Nasal Spray 10 mcg/0.1 mL, and Stimate® Nasal Spray 1.5 mg/mL.
About this Recall:
Ferring Pharmaceuticals US is voluntarily recalling all lots on the market of DDAVP nasal spray 10 mcg/0.1mL, desmopressin acetate nasal spray 10 mcg/0.1mL, and Stimate® nasal spray 1.5 mg/mL. These products are being recalled due to amounts of desmopressin higher than specified. These results were obtained during routine testing.
What this means to you:
The risks associated with higher than specified amounts of desmopressin are due to abnormally low levels of sodium in the blood (e.g., hyponatremia) which could eventually lead to seizure, coma, and death. To date, Ferring has not received an increase in adverse event reports due to increased concentrations of desmopressin from users of the nasal spray. However, a single non-fatal adverse event potentially associated with this issue was reported during the timeframe that the impacted product was distributed.
DDAVP nasal spray and desmopressin nasal spray are both indicated as antidiuretic replacement therapy in the management of central cranial diabetes insipidus as well as for the management of temporary polyuria and polydipsia following head trauma or surgery in the pituitary region. Stimate nasal spray is indicated for the treatment of patients with hemophilia A with Factor VIII coagulant activity levels greater than 5% and for the treatment of patients with mild to moderate classic von Willebrand’s disease (Type I) with Factor VIII levels greater than 5%.
The impacted product names, including the batch numbers and expiration dates, are available on the FDA Recall Notification, listed at the link on the following page. Patients and consumers can contact their physician or healthcare provider with any questions related to this recall.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Ferring Issues Voluntary Nationwide Recall of DDAVP® Nasal Spray 10 mcg/0.1 mL, Desmopressin Nasal Spray 10 mcg/0.1 mL, Stimate® Nasal Spray 1.5 mg/mL Due to Superpotency
FDA Publish Date: 08/05/2020
Ferring Pharmaceuticals is voluntarily recalling all lots on the market of DDAVP® Nasal Spray 10 mcg/0.1 mL, Desmopressin Acetate Nasal Spray 10 mcg/0.1 mL, and Stimate® Nasal Spray 1.5 mg/mL listed in the table on the following page to the consumer level. These products are being recalled due to superpotency or amounts of desmopressin higher than specified. These out of specification results were obtained during routine testing.
The risks associated with higher than specified amounts of desmopressin relate to abnormally low levels of sodium in the blood (e.g., hyponatremia) which could eventually lead to seizure, coma, and death. To date, Ferring has not received an increase in adverse event reports due to increased concentrations of desmopressin from users of the nasal spray. A single non-fatal adverse event potentially associated with this issue was reported in the US during the timeframe that the affected product was distributed.
DDAVP nasal spray and desmopressin nasal spray are both indicated as antidiuretic replacement therapy in the management of central cranial diabetes insipidus as well as for the management of temporary polyuria and polydipsia following head trauma or surgery in the pituitary region. Stimate nasal spray is indicated for the treatment of patients with hemophilia A with Factor VIII coagulant activity levels greater than 5% and for the treatment of patients with mild to moderate classic von Willebrand’s disease (Type I) with Factor VIII levels greater than 5%.
Ferring is notifying its distributors and wholesale customers by letter and asking them to check for impacted product and to return unused product through directions provided in the recall letter. The affected product name, including the batch numbers and expiration dates are found in the table below.
|
Lot |
Expiration Date |
NDC # |
|
DDAVP® Nasal Spray 10 mcg/0.1 mL, 5 mL |
||
|
N14695F |
08-2020 |
55566-2500-00 |
|
N15627C |
10-2020 |
55566-2500-00 |
|
P11319P |
01-2021 |
55566-2500-00 |
|
P11706F |
04-2021 |
55566-2500-00 |
|
R11842C |
03-2022 |
55566-2500-00 |
|
R13637E |
06-2022 |
55566-2500-00 |
|
Desmopressin Acetate Nasal Spray 10 mcg/0.1 mL, 5 mL |
||
|
N14695P |
08-2020 |
69918-0501-05 |
|
N14695S |
08-2020 |
69918-0501-05 |
|
N15627G |
10-2020 |
69918-0501-05 |
|
N15627GA |
10-2020 |
69918-0501-05 |
|
P10422A |
01-2021 |
69918-0501-05 |
|
P10422AA |
01-2021 |
69918-0501-05 |
|
P10430G |
03-2021 |
69918-0501-05 |
|
P11319M |
01-2021 |
69918-0501-05 |
|
P12969H |
05-2021 |
69918-0501-05 |
|
P12969IR |
05-2021 |
69918-0501-05 |
|
P13216G |
05-2021 |
69918-0501-05 |
|
P13216P |
05-2021 |
69918-0501-05 |
|
R11842A |
03-2022 |
69918-0501-05 |
|
R11842S |
03-2022 |
69918-0501-05 |
|
R12630A |
05-2022 |
69918-0501-05 |
|
R13071H |
04-2022 |
69918-0501-05 |
|
Stimate® Nasal Spray 1.5 mg/mL, 2.5 mL |
||
|
N14134C |
07-2020 |
00053-6871-00 |
|
N15378G |
09-2020 |
00053-6871-00 |
|
N17445N |
12-2020 |
00053-6871-00 |
|
P11326AA |
02-2021 |
00053-6871-00 |
|
P11326C |
02-2021 |
00053-6871-00 |
|
P13209L |
04-2021 |
00053-6871-00 |
|
P13212H |
06-2021 |
00053-6871-00 |
|
P13755A |
06-2021 |
00053-6871-00 |
|
P13756P |
08-2021 |
00053-6871-00 |
|
R11845A |
04-2022 |
00053-6871-00 |
|
R13271A |
04-2022 |
00053-6871-00 |
|
R13648A |
06-2022 |
00053-6871-00 |
|
R14101A |
07-2022 |
00053-6871-00 |
|
R14667A |
08-2022 |
00053-6871-00 |
|
R15953C |
09-2022 |
00053-6871-00 |
Distributors/wholesalers with questions regarding the recall should call Stericycle at 1-888-228-5053.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail or by fax.
- Complete and submit the report Online.
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
This recall is being conducted with the knowledge of the US Food and Drug Administration.
About Ferring Pharmaceuticals
Ferring Pharmaceuticals is a research-driven, specialty biopharmaceutical group committed to helping people around the world build families and live better lives. In the United States, Ferring is a leader in reproductive medicine and maternal health, and in specialty areas within gastroenterology and orthopedics. For more information, call 1-888-FERRING (1-888-337-7464); visit www.FerringUSA.com.
Link to FDA recall notification
Company Contact Information
Consumers:
- 1-888-337-7464
Media: Patrick Gorman
- (862) 286-5035
- patrick.gorman@ferring.com
Product Photos
- Click here for product photos.
Recall of Metformin ER Tablets, 500 mg and 1,000 mg, by Lupin Pharmaceuticals
Date: 7/8/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Lupin Pharmaceuticals has posted a market recall of metformin extended-release (ER) tablets, 500 mg and 1,000 mg.
About this Recall:
Lupin Pharmaceuticals is voluntarily recalling all batches of Metformin ER Tablets, 500 mg and 1,000 mg to the consumer level. As part of the ongoing evaluation and communication with the Food & Drug Administration (FDA), additional testing showed that certain tested batches were above the acceptable daily intake (ADI) limit for the impurity N-Nitrosodimethylamine (NDMA). Out of an abundance of caution, the company is recalling all batches of metformin ER tablets in the strengths of 500 mg and 1,000 mg in the United States (US).
What this means to you:
NDMA is a substance that could cause cancer based on results from laboratory tests. NDMA is a known environmental contaminant and is found in water and foods (including meats, dairy products, and vegetables). Metformin ER tablets are a prescription oral medication used as an adjunct to diet and exercise to improve blood glucose control in adults with type 2 diabetes mellitus. The recalled NDCs are available at the FDA Recall Notification link found in the box on the following page.
Patients taking recalled metformin ER tablets, 500 mg or 1,000 mg, are advised to continue taking their medication and contact their pharmacist, physician, or medical provider for advice regarding an alternative treatment. According to the US FDA, it could be dangerous for patients with this serious condition to stop taking their metformin without first talking to their healthcare providers. Please visit the agency’s website for more information.
Consumers with questions regarding this recall should contact Inmar Rx Solutions at (855) 532-1856 Monday through Friday 9:00 AM to 5:00 PM EST. For reimbursement, please have the recalled NDCs returned to Inmar Rx Solutions; the NDC number can be found on the top of the bottle label.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online
or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Lupin Pharmaceuticals Issues Voluntarily Nationwide Recall of Metformin ER Tablets, 500 mg and 1,000 mg Due to the Detection of NDMA Impurity
FDA Publish Date: 7/8/2020
Lupin Pharmaceuticals is voluntarily recalling all batches of Metformin Hydrochloride (HCl) Extended-Release (ER) Tablets USP, 500 mg and 1000 mg to the consumer level. As part of the ongoing assessment and continuation of dialog with the Food & Drug Administration (FDA), additional analysis revealed that certain tested batches were above the acceptable daily intake (ADI) limit for the impurity N-Nitrosodimethylamine (NDMA). Out of an abundance of caution, the company is recalling all batches of Metformin HCl ER Tablets USP, 500 mg and 1,000 mg in the United States (US). To date, Lupin Pharmaceuticals has not received any reports of adverse events related to this recall.
Risk Statement: NDMA is classified as a probable human carcinogen (a substance that could cause cancer) based on results from laboratory tests. NDMA is a known environmental contaminant and is found in water and foods, including meats, dairy products, and vegetables.
Metformin ER is a prescription oral medication indicated as an adjunct to diet and exercise to improve blood glucose control in adults with type 2 diabetes mellitus. Metformin ER tablets 500 mg and 1,000 mg are packaged in 60, 90, and 100 count bottles and were distributed nationwide in the US to wholesalers, distributors, drug chain pharmacies, mail order pharmacies, and supermarkets. The recalled NDCs are included in the table below:
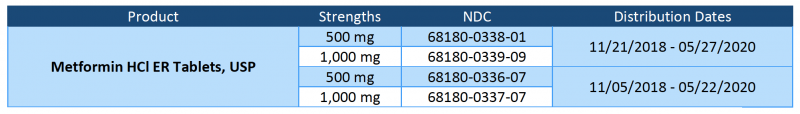
Lupin Pharmaceuticals is notifying its wholesalers, distributors, drug chain pharmacies, mail order pharmacies, and supermarket customers by phone and through recall notification and is arranging for the return of all the recalled product NDCs.
Patients taking Metformin HCl ER Tablets, USP 500 mg or 1,000 mg, are advised to continue taking their medication and contact their pharmacist, physician, or medical provider for advice regarding an alternative treatment. According to the US FDA, it could be dangerous for patients with this serious condition to stop taking their metformin without first talking to their healthcare professionals. Please visit the agency’s website for more information at https://www.fda.gov/drugs/drug-safety-and-availability/fda-updates-and-press-announcements-ndma-metformin.
Wholesalers, distributors, and retailers that have Metformin HCl ER Tablets USP, 500 mg and 1,000 mg that are being recalled should discontinue distribution of the recalled product NDCs immediately and return it to Inmar Rx Solutions, 635 Vine St., Winston Salem, NC 27101 (phone 855-532-1856).
Consumers, wholesalers, distributors, and retailers with questions regarding this recall should contact Inmar Rx Solutions, Inc. at (855) 532-1856 Monday through Friday 9:00 AM to 5:00 PM EST. For reimbursement, please have the recalled NDCs returned to Inmar Rx Solutions; the NDC number can be found on the top of the bottle label.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail or by fax.
• Complete and submit the report Online
• Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the US FDA.
Link to FDA recall notification
Company Contact Information
Consumers: Inmar, Rx Solutions, Inc.
- (855) 532-1856
Media: Arvind Bothra
- arvindbothra@lupin.com
Product Photos
FDA Recall of Metformin ER Tablets, 750 mg by Granules Pharmaceuticals
Date: 7/6/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Granules Pharmaceuticals has posted a lot recall of metformin extended-release (ER) tablets, 750 mg.
About this recall:
Granules Pharmaceuticals is voluntarily recalling twelve (12) lots of Metformin ER Tablets, 750 mg, supplied in 100 and 500 count bottles within expiration to the consumer level due to the detection of N-Nitrosodimethylamine (NDMA) levels above the acceptable daily intake (ADI) limit.
Granules’ test results showed NDMA levels above the FDA’s acceptable limit in one (1) out of the twelve (12) batches distributed in the United States (US). All other batches continue to remain within the required limits. Out of abundance of caution, Granules Pharmaceuticals has decided to voluntarily recall all twelve (12) of the distributed lots within expiration of metformin ER tablets, 750 mg.
Granules’ metformin immediate-release (IR) tablets in the strengths of 500 mg, 850 mg, and 1,000 mg as well as their metformin ER tablets in the 500 mg strength are not impacted by this recall.
What this means to you:
NDMA is a substance that could cause cancer based on results from laboratory tests. NDMA is a known environmental contaminant and is found in water and foods (including meats, dairy products, and vegetables).
Metformin ER tablets, 750 mg, are used as an adjunct to diet and exercise to improve blood sugar control in adults with type 2 diabetes mellitus. Details for identifying the recalled lots are available at the FDA Recall Notification link found on the following page.
Patients with questions regarding this recall or wishing to return product may contact Inmar Pharmaceutical Services recall processor for instructions and a return kit for returning their medication:
- Contact Inmar at 888-985-9117 (hours of operation: 9 AM to 5 PM Eastern Time, Monday through Friday) or email Inmar at: rxrecalls@inmar.com
- Inmar will provide the materials needed to return medication and instructions for reimbursement
If you would like to report any adverse reactions or quality problems experienced with the use of this product you may contact Granules Drug Safety by phone at 1-877-770-3183 Monday through Friday, 8:00 AM to 8:00 PM EST, or via email at drugs.safety@granulesindia.com.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Granules Pharmaceuticals Issues Voluntary Nationwide Recall of Metformin ER Tablets USP, 750 mg Due to the Detection of NDMA Impurity
FDA Publish Date: 07/06/2020
Granules Pharmaceuticals, Inc., is voluntarily recalling twelve (12) lots of Metformin Hydrochloride (HCl) Extended-Release (ER) Tablets USP, 750 mg, 100 and 500 count bottles within expiry to the consumer level due to the detection of N-Nitrosodimethylamine (NDMA) levels above the acceptable daily intake limit.
Granules’ test results showed NDMA levels above the FDA’s acceptable limit in one (1) out of the twelve (12) batches distributed to the United States (US) market. All other batches continue to remain within the specifications. Out of abundance of caution, Granules Pharmaceuticals has decided to voluntarily recall all twelve (12) of the distributed lots within expiry of Metformin HCl ER Tablets USP, 750 mg from the market.
Granules Pharmaceuticals, Inc. has not received any reports of adverse events that have been confirmed to be directly related to this recall as of the date of this letter.
Granules’ Metformin HCl Immediate-Release (IR) Tablets USP, 500 mg, 850 mg, & 1,000 mg and Metformin HCl ER Tablets USP, 500 mg are not affected by this recall.
Risk Statement: NDMA is classified as a probable human carcinogen (a substance that could cause cancer) based on results from laboratory tests. NDMA is a known environmental contaminant and found in water and foods, including meats, dairy products, and vegetables.
Metformin HCl ER Tablets USP, 750 mg are indicated as an adjunct to diet and exercise to improve blood sugar control in adults with type 2 diabetes mellitus. The metformin ER tablets, 750 mg, lots subject to the recall are identified in the table on the following page.
Metformin HCl ER Tablets USP, 750 mg
|
NDC |
Bottle Count |
Lot/Expiration |
|
70010-0492-01 |
100 count bottles |
4920003A/May-21 4920004A/Jun-21 4920005A/Jun-21 4920009A/Nov-21 4920010A/May-22 4920011A/Jun-22 4920012A/Jun-22 4920013A/Jul-22 4920014A/Jul-22 4920015A/Aug-22 4920016A/Jan-23 |
|
70010-0492-05 |
500 count bottles |
4920005B/Jun-21 |
The affected metformin ER tablets, 750 mg, lots were distributed nationwide in the US directly to distributors and retailers. Granules Pharmaceuticals is in the process of notifying its distributers and customers affected by this recall via mail (FedEx standard overnight) by mailing a recall notification letter and is arranging for return of the entire recalled product. Anyone with an existing inventory of the product should quarantine the recalled lots immediately.
Customers and patients with questions regarding this recall or wishing to return product may contact Inmar Pharmaceutical Services product recall processor to obtain instructions and a return kit for returning their medication:
• Contact Inmar at 888-985-9117 (hours of operation: 9 AM to 5 PM Eastern Time, Monday through Friday) or email Inmar at: rxrecalls@inmar.com
• Inmar will provide the materials needed to return their medication and instructions for reimbursement
If you would like to report any adverse reactions or quality problems experienced with the use of this product you may contact Granules Drug Safety by phone at 1-877-770-3183 Monday through Friday, 8:00 AM to 8:00 PM EST, or via email at drugs.safety@granulesindia.com.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the US Food and Drug Administration (FDA).
Link to FDA Recall Notification
Company Contact Information
Consumers: Inmar, Pharmaceutical Services
- 888-985-9117
- rxrecalls@inmar.com
Media: Mary D Villalona, Director, Quality
- 571-665-2914
- mary.villalona@granulespharma.com
Product Photos
Click here to view product photos.
Children’s Robitussin® Honey Cough and Chest Congestion DM and Children’s Dimetapp® Cold and Cough Recall
Date: 06/18/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
GSK Consumer Healthcare has posted a lot recall of Children’s Robitussin® Honey Cough and Chest Congestion DM and Children’s Dimetapp® Cold and Cough.
About this Recall:
GSK Consumer Healthcare is voluntarily recalling to the retail level two (2) lots of Children’s Robitussin® Honey Cough and Chest Congestion DM and one (1) lot of Children’s Dimetapp® Cold and Cough (listed at the FDA Recall Notification link below), as the packages contain incorrect dosing cups. During the review of the packaging for these products, GSK discovered that the dosing cups for the Children’s Robitussin® Honey product are missing the 5 mL and 10 mL graduations, and the dosing cups for the Children’s Dimetapp® product are missing the 10 mL graduation. The dosing cups packaged with both products only have the 20 mL graduation.
What this means to you:
There is a potential risk of accidental overdose if caregivers dispensing the syrup do not notice the differences between the graduations printed on the dosing cups and the indicated amounts to be administered (as directed in the Instructions for Use). Children’s Robitussin Honey Cough & Chest Congestion DM contains 10 mg dextromethorphan and guaifenesin 100 mg per 10 mL and is labeled for children 4 years and older and for adults. Children’s Dimetapp Cold & Cough contains 2 mg brompheniramine, 10 mg dextromethorphan, and 5 mg phenylephrine per 10 mL, and is labeled for children 6 years and older and adults. Symptoms of overdose of either product may include any of the following: impaired coordination; increase or decrease in energy; elevation in blood pressure, heart rate, or respiration; severe dizziness or drowsiness; slow heart rate; fainting; restlessness; seizure; decreased respiration; nausea; vomiting; constipation; diarrhea; abdominal pain; visual and hearing hallucinations; and urinary retention. As of the date of the recall, GSK had not received any adverse events related to this recall or consumer complaints about the incorrect dosing cups supplied with the product.
Consumers with questions regarding this recall or to report an adverse experience please call 1-800-762-4675, Monday through Friday, 8:00 AM – 6:00 PM EST.
Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this product.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
GSK Consumer Healthcare Issues Voluntary Nationwide Recall of Children’s Robitussin® Honey Cough and Chest Congestion DM and Children’s Dimetapp® Cold and Cough Due to Dosing Cups Missing Some Graduation Markings
FDA Publish Date: 06/18/2020
GSK Consumer Healthcare is voluntarily recalling to the retail level two (2) lots (listed below) of Children’s Robitussin® Honey Cough and Chest Congestion DM and one (1) lot of Children’s Dimetapp® Cold and Cough, due to the inclusion of incorrect dosing cups. During the review of the packaging documents for these products, GSK discovered that the dosing cups for the Children’s Robitussin® Honey product are missing the 5 mL and 10 mL graduations, while the dosing cups for the Children’s Dimetapp® product are missing the 10 mL graduation. The dosing cups packaged with both products only have the 20 mL graduation.
There is a potential risk of accidental overdose if caregivers dispensing the syrup do not notice the discrepancies between the graduations printed on the dosing cups and the indicated amounts to be administered (as directed in the Instructions for Use). Children’s Robitussin Honey Cough & Chest Congestion DM contains 10 mg dextromethorphan HBr USP and guaifenesin USP 100 mg per 10 mL, and is labeled for children 4 and older, as well as adults. Children’s Dimetapp Cold & Cough contains 2 mg brompheniramine maleate USP, 10 mg dextromethorphan HBr USP, and 5 mg phenylephrine HCl USP per 10 mL, and is labeled for children 6 and older, as well as adults. Symptoms of overdose of either product may include any of the following: impaired coordination; brain stimulation causing increase in energy, elevation in blood pressure, heart rate, and respiration; a lack of energy and enthusiasm; severe dizziness or drowsiness; slow heart rate; fainting; psychotic behavior; restlessness; seizure; decreased respiration; nausea; vomiting; constipation; diarrhea; abdominal pain; visual and hearing hallucinations; urinary retention. As of the date of the recall announcement, GSK Consumer Healthcare has not received any adverse events related to these products or consumer complaints regarding the incorrect dosing cups supplied with the product.
The recall is limited to the three (3) lots listed below:
|
Drug Name |
NDC |
Lots (Expiration) |
|
Children’s Robitussin® Honey Cough and Chest Congestion DM (4 oz) |
00031-8760-12 |
02177 (Jan 2022) 02178 (Jan 2022) |
|
Children’s Dimetapp® Cold and Cough (8 oz) |
00031-2234-19 |
CL8292 (Sep 2021) |
These lots were distributed nationwide between February 5, 2020 and June 3, 2020 within the United States (US). GSK Consumer Healthcare has notified wholesalers, distributors, and retailers to arrange for return of any recalled product. Wholesalers, distributors, and retailers with an existing inventory of the lots being recalled should stop distribution and quarantine these lots immediately. Wholesalers, distributors, and retailers that have further distributed the recalled product should notify any accounts or additional locations which may have received the recalled product from them.
Consumers with questions regarding this recall or to report an adverse experience please call 1-800-762-4675, Monday through Friday, 8:00 AM – 6:00 PM EST.
Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the US Food and Drug Administration (FDA).
Company Contact Information
Consumers: GSK Contact Center
- 1-800-762-4675
Product Photos
For photos of this recall, please refer to the FDA link.
Metformin ER 500 mg Tablets
Date: 06/11/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Lupin Pharmaceuticals has posted a recall of Metformin Extended-Release (ER) Tablets, 500 mg.
About this Recall:
Lupin Pharmaceuticals is voluntarily recalling 1 lot of Metformin ER Tablets, 500 mg, to the consumer level.
FDA testing showed that this lot had higher levels than the Acceptable Daily Intake limit for the impurity N-Nitrosodimethylamine (NDMA).
What this means to you:
NDMA is a substance that could cause cancer based on results from laboratory tests. NDMA is a known environmental contaminant and is found in water and foods (such as meats, dairy products, and vegetables).
The FDA Recall Notification linked below lists the recalled NDC and lot number. The product can be identified by the NDC and lot number listed on the side of the bottle label. Metformin ER tablets were distributed nationwide in the United States (US) to wholesalers, distributors, and mail order pharmacies.
Patients with recalled metformin ER tablets are advised by the manufacturer to continue taking their medication and contact their pharmacist, physician, or medical provider for advice regarding an alternative treatment. According to the US Food & Drug Administration (FDA), it could be dangerous for patients with this serious condition to stop taking their metformin without first talking to their healthcare provider.
Consumers with questions about this recall should contact Inmar Rx Solutions at (855) 532-1856, Monday through Friday, 9:00 AM to 5:00 PM EST. For reimbursement, please have the recalled lot returned to Inmar Rx Solutions; the lot number can be found on the side of the bottle.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Lupin Issues Voluntary Nationwide Recall of Metformin ER Tablets, 500 mg, Due to the Detection of NDMA
FDA Publish Date: 06/11/2020
Lupin Pharmaceuticals Inc. is voluntarily recalling Metformin Hydrochloride Extended-Release (ER) Tablets USP (generic equivalent of Fortamet®), 500 mg, lot G901203, to the consumer level. FDA analysis revealed that this lot exceeded the Acceptable Daily Intake (ADI) limit for the impurity N-Nitrosodimethylamine (NDMA). To date, Lupin has not received any reports of adverse events related to this recall.
Risk Statement: NDMA is classified as a probable human carcinogen (a substance that could cause cancer) based on results from laboratory tests. NDMA is a known environmental contaminant and found in water and foods (such as meats, dairy products, and vegetables).
Metformin ER is a prescription oral medication indicated as an adjunct to diet and exercise to improve blood glucose control in adults with type 2 diabetes mellitus. The product is packaged in a bottle containing 60 tablets with NDC 68180-0336-07. The impacted lot of metformin ER tablets 500 mg is listed in the below table:
|
Product Name |
NDC |
Lot Number |
Expiration Date |
|
Metformin Hydrochloride Extended-Release Tablets USP, 500mg |
68180-0336-07 |
G901203 |
12/2020 |
Lupin is notifying these wholesalers, distributors, and mail order pharmacies by phone and through recall notification and is arranging for the return of all the recalled product. The manufacturer is advising patients taking recalled product to continue taking their medication and contact their pharmacist, physician, or medical provider for advice regarding an alternative treatment. According to the US Food & Drug Administration (FDA), it could be dangerous for patients with this serious condition to stop taking their metformin without first talking to their healthcare professionals. Please visit the agency’s website for more information.
Wholesalers, distributors, and retailers that have metformin ER 500 mg tablets which are being recalled should discontinue distribution of the recalled product lot immediately and return it to Inmar Rx Solutions, 635 Vine St., Winston Salem, NC 27101, phone (855) 532-1856.
Consumers, wholesalers, distributors, and retailers with questions regarding this recall should contact Inmar Rx Solutions, Inc. at (855) 532-1856 Monday through Friday 9:00 AM to 5:00 PM EST. For reimbursement, please have the recalled lot returned to Inmar Rx Solutions; the lot number can be found on the side of the bottle.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail or by fax.
- Complete and submit the report online
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the US Food and Drug Administration.
Company Contact Information
Consumers: Inmar Rx Solutions, Inc.
- (855) 532-1856
Media: Arvind Bothrar
- +91-22-66402137
- arvindbothra@lupin.com
Product Photos
For product photos, please refer to the FDA Link.
Metformin ER 500 mg Tablets
Date: 06/01/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Amneal has posted a recall of Metformin Hydrochloride Extended-Release (ER) Tablets, 500 mg and 750 mg.
About this Recall:
Amneal Pharmaceuticals is voluntarily recalling all lots of Metformin ER Tablets, 500 mg and 750 mg, within expiry to the retail level.
Amneal was notified by the United States (US) Food and Drug Administration (FDA) that the agency’s testing of seven lots of Metformin ER Tablets, 500 mg and 750 mg, showed N-Nitrosodimethylamine (NDMA) amounts above acceptable FDA levels. FDA recommended the recall of the seven tested lots. Amneal has agreed to this recall and has further decided to extend the recall to all lots within expiry of Metformin Hydrochloride ER Tablets, 500 mg and 750 mg, to be extra careful.
What this means to you:
NDMA is a substance that could cause cancer based on results from laboratory tests. NDMA is a known environmental contaminant and is found in water and foods (such as meats, dairy products, and vegetables). The impacted metformin ER tablets can be identified by the NDC number found on the product label. The recalled NDC number is listed on the FDA Recall Notification at the link below. The FDA has stated patients should continue taking metformin tablets, even after recalls occur, until they speak with their healthcare provider who can prescribe a replacement.
Amneal is notifying its direct customers via mail (UPS Standard Overnight) by mailing a recall notification letter and is arranging for return of all the recalled product. Anyone with an existing inventory of the product should quarantine the recalled lots immediately.
Customers who purchased the impacted product directly from Amneal may call Amneal at 1-833-582-0812 or email to AmnealproductrecallDS@amneal.com, Monday – Friday, 8:00 am – 5:00 pm, EST, for further information.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Amneal Pharmaceuticals LLC Issues Voluntary Nationwide Recall of Metformin Hydrochloride Extended Release Tablets, USP, 500 mg and 750 mg, Due to the Detection of NDMA
FDA Publish Date: 06/01/2020
Amneal Pharmaceuticals LLC Bridgewater, New Jersey (Amneal), is voluntarily recalling all lots of Metformin Hydrochloride Extended Release Tablets, USP, 500 mg and 750 mg, within expiry to the Retail Level.
Amneal was notified by the U.S. FDA that the Agency’s testing of seven lots of Metformin Hydrochloride Extended Release Tablets, USP, 500 mg and 750 mg, showed N-Nitrosodimethylamine (NDMA) amounts above acceptable FDA levels. FDA recommended the recall of the seven tested lots. Amneal has agreed to this recall and has further decided to extend the recall to all lots within expiry of Metformin Hydrochloride Extended Release Tablets, USP, 500 mg and 750 mg, out of an abundance of caution. Further scientific evaluations are ongoing at Amneal.
To date, Amneal has not received any reports of adverse events that have been confirmed to be directly related to this recall.
Amneal’s Metformin Hydrochloride Immediate Release Tablets, USP are not affected by this recall.
Risk Statement: NDMA is classified as a probable human carcinogen (a substance that could cause cancer) based on results from laboratory tests. NDMA is a known environmental contaminant found in water and foods, including meats, dairy products and vegetables.
Metformin HCl Extended Release Tablets, USP, 500 mg and 750 mg, manufactured by Amneal, are prescription, solid oral products that are indicated as an adjunct to diet and exercise to improve blood sugar control in adults with type 2 diabetes mellitus.
The Metformin Hydrochloride Extended Release Tablets, USP, 500 mg and 750 mg, subject to the recall, are identified by the NDC numbers stated on the product label.
Metformin HCl Extended Release Tablets, USP, 500 mg
|
|
|
|
53746-178-01 |
100 count bottles |
|
53746-178-05 |
500 count bottles |
|
53746-178-10 |
1000 count bottles |
|
53746-178-90 |
90 count bottles |
|
53746-178-Bulk |
Bulk Box |
|
65162-178-09 |
90 count bottles |
|
65162-178-10 |
100 count bottles |
|
65162-178-11 |
1000 count bottles |
|
65162-178-50 |
500 count bottles |
Metformin HCl Extended Release Tablets, USP, 750 mg
|
|
|
|
53746-179-01 |
1000 count bottles |
|
53746-179-Bulk |
Bulk Box |
|
65162-179-10 |
100 count bottles |
The affected Metformin Hydrochloride Extended Release Tablets, USP, 500 mg and 750 mg, lots were distributed nationwide in the US directly to Wholesalers, Distributors, Retailers and Repackagers.
Amneal is notifying its direct customers via mail (UPS Standard Overnight) by mailing a recall notification letter and is arranging for return of all the recalled product. Anyone with an existing inventory of the product should quarantine the recalled lots immediately.
Customers who purchased the impacted product directly from Amneal may call Amneal at 1-833-582-0812 or email to AmnealproductrecallDS@amneal.com, Monday – Friday, 8:00 am – 5:00 pm, EST, for further information.
Retailers who have Metformin Hydrochloride Extended Release Tablets, USP, 500 mg or 750 mg, which are being recalled, should cease dispensing and contact Inmar at 855-532-1851 or via email at Rxcalls@inmar.com Monday – Friday, 8:00 am – 5:00 pm, EST, to arrange for product return.
If you would like to report any adverse reactions or quality problems experienced with the use of this product you may contact Amneal Drug Safety by phone at 1-877-835-5472, Monday – Friday, 8:00 am – 6:00 pm, EST, or via e-mail at DrugSafety@amneal.com.
Any adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail or by fax.
- Complete and submit the report online
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the U.S. Food and Drug Administration.
Company Contact Information
Consumers:
Amneal
1-833-582-0812
AmnealproductrecallDS@amneal.com
Media:
Candis Edwards
Product Photos
For product photos please refer to the FDA Link
Metformin ER 500 mg Tablets
Date: 05/28/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Apotex has posted a recall of Metformin Hydrochloride Extended-Release (ER) Tablets, 500 mg.
About this Recall:
Apotex is voluntarily recalling all lots of Metformin Hydrochloride (HCl) ER Tablets, 500 mg within expiration to the retail level. The United States (US) Food and Drug Administration (FDA) tested 1 lot of Apotex’s metformin HCl ER tablets, and found that tablets contain levels of the N-Nitrosodimethylamine (NDMA) impurity greater than the acceptable daily intake limit. As a result, the FDA recommended recall of the 1 tested lot. Apotex is recalling this lot, and out of an abundance of caution, the company has extended the recall to include all lots of Apotex’s metformin ER tablets in the US. Selling of this product was stopped in the US in February 2019 and only limited product remains on the market. To date, there have not been any reports of adverse events related to use of this product.
What this means to you:
NDMA is a substance that could cause cancer based on results from laboratory tests. NDMA is a known environmental contaminant and is found in water and foods (such as meats, dairy products, and vegetables). The impacted metformin ER tablets can be identified by the NDC number found on the product label. The recalled NDC number is listed on the FDA Recall Notification at the link below. The FDA has stated patients should continue taking metformin tablets, even after recalls occur, until they speak with their healthcare provider who can prescribe a replacement.
Consumers with questions regarding this recall can contact Apotex by phone at 1-800-706-5575 (8:30 AM – 5:00 PM, EST Monday through Friday) or email UScustomerservice@Apotex.com. Consumers should contact their physician or health-care provider if they have experienced any problems that may be related to taking or using this drug product.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Apotex Issues Voluntary Nationwide Recall of Metformin ER Tablets, 500 mg Due to the Detection of NDMA
FDA Publish Date: 05/28/2020
Apotex Corp. is voluntarily recalling all lots of Metformin Hydrochloride (HCl) ER Tablets, USP 500 mg within expiration to the retail level. The United States (US) Food and Drug Administration (FDA) tested 1 lot of Apotex’s metformin HCl ER tablets, and it was found to contain levels of the N-Nitrosodimethylamine (NDMA) impurity greater than the acceptable daily intake limit. As a result, the FDA recommended recall of the 1 tested lot. Apotex is recalling this lot, and out of an abundance of caution, the company has extended the recall to include all lots of Apotex’s metformin HCl ER tablets in the US. Selling of this product was stopped in the US in February 2019 and only limited product remains on the market. To date, no reports of adverse events related to use of this product have been received.
Risk Statement: NDMA is classified as a substance that could cause cancer based on results from laboratory tests. NDMA is a known environmental contaminant and is found in water and foods (such as meats, dairy products, and vegetables). The FDA has stated patients should continue taking metformin tablets, even after recalls occur, until they speak with their health care provider who can prescribe a replacement.
|
Product |
Strength. |
Pack Size |
NDC Number |
|
Metformin Hydrochloride Extended-Release Tablets, USP |
500 mg |
100’s bottle |
60505-0260-01 |
The impacted metformin ER tablets were distributed nationwide in the US to warehouse chains. Apotex is in the process of notifying its affected direct account wholesaler, distributor, chain distribution, and warehousing chains via mail (FedEx Standard Overnight) by mailing a recall notification letter and is arranging for return of all recalled product.
Wholesalers, distributors and retailers should return the recalled product to the place of purchase. Anyone with an existing inventory of the product should quarantine the recalled lots immediately. Customers who purchased the impacted product directly from Apotex can call Inmar Rx Solutions at 1-888-985-9014 (option 1) (9:00 AM – 5:00 PM, EST Monday through Friday), to arrange for their return.
2—0528-2020 Class 2 Metformin ER 500 mg – Apotex Corp, Voluntary Nationwide Recall
Consumers with questions regarding this recall can contact Apotex by phone at 1-800-706-5575 (8:30 AM – 5:00 PM, EST Monday through Friday) or email UScustomerservice@Apotex.com. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report online
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the US Food and Drug Administration (FDA).
Company Contact Information
Consumers:
- Apotex Corp.
- 1-800-706-5575
- UScustomerservice@Apotex.com
Media:
- Jordan Berman
- 1 (416) 749-9026 Ext. 7487
- jberman@apotex.com
Product photos

NP Thyroid® (Thyroid Tablets, USP)
Date: 05/22/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Acella Pharmaceuticals, LLC has posted a lot recall of NP Thyroid® (Thyroid Tablets, USP) 30 mg, 60 mg, and 90 mg.
About this Recall:
Acella Pharmaceuticals, LLC is voluntarily recalling a total of 13 lots of 30 mg, 60 mg, and 90 mg NP Thyroid® (thyroid tablets, USP) to the consumer level. The products are being recalled because our testing has found these lots to be superpotent. The product may have up to 115% of the labeled amount of liothyronine (T3).
What this means to you:
Patients being treated for hypothyroidism (underactive thyroid), who receive superpotent NP Thyroid, may experience signs and symptoms of hyperthyroidism (overactive thyroid) which include, but are not limited to, weight loss, heat intolerance, fatigue, muscle weakness, high blood pressure, chest pain, rapid heart rate, or heart rhythm disturbances. Pregnant women who take superpotent NP Thyroid may also experience negative maternal and fetal outcomes including miscarriage and/or impairment of fetal development. The FDA is recommending patients who are currently taking NP Thyroid from a lot being recalled should not discontinue use without contacting their healthcare provider for further guidance and/or a replacement prescription. View the FDA Recall Notification at the link below for a listing of lots being recalled.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Acella Pharmaceuticals, LLC Issues Voluntary Nationwide Recall of Certain Lots of NP Thyroid® (Thyroid Tablets, USP) Due to Super Potency
FDA Publish Date: 05/22/2020
Acella Pharmaceuticals, LLC is voluntarily recalling a total of 13 lots of 30 mg, 60 mg, and 90 mg NP Thyroid® (thyroid tablets, USP) to the consumer level. The products are being recalled because testing has found these lots to be superpotent. The product may have up to 115% of the labeled amount of Liothyronine (T3).
Risk Statement: Patients being treated for hypothyroidism (underactive thyroid), who receive superpotent NP Thyroid, may experience signs and symptoms of hyperthyroidism (overactive thyroid) which include, but are not limited to, weight loss, heat intolerance, fatigue, muscle weakness, hypertension (high blood pressure), chest pain, rapid heart rate, or heart rhythm disturbances. Pregnant women who take superpotent NP Thyroid may also experience negative maternal and fetal outcomes including miscarriage and/or impairment of fetal development. The FDA is recommending patients talk to their healthcare professional before they stop taking their NP Thyroid medicine. To date, Acella has received 2 reports of adverse events known to be related to this recall.
NP Thyroid (thyroid tablets, USP) is composed of levothyroxine and liothyronine, and used to treat hypothyroidism (underactive thyroid). The products subject to recall are packed in 100-count bottles. Product images are provided below.
To best identify the product, the NDCs, product description, lot numbers, and expiration dates are listed. These lots were distributed nationwide in the United States (US) to Acella’s direct accounts.
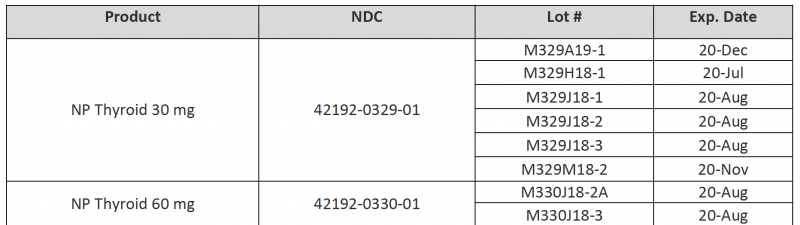
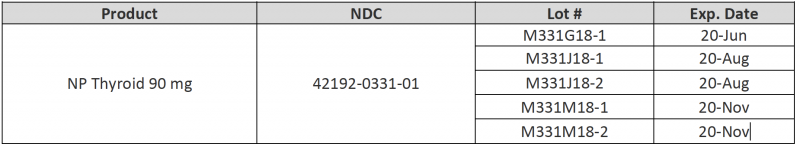
Acella is proactively notifying its wholesalers by email and phone to discontinue distribution of the product being recalled and is arranging for return of all recalled products. Patients who are currently taking NP Thyroid from the lots being recalled should not discontinue use without contacting their healthcare provider for further guidance and/or a replacement prescription.
Consumers with questions about the recall can email Acella Pharmaceuticals at recall@acellapharma.com or contact Acella Customer Service at 1-800-541-4802, Monday through Thursday from 9:00 am to 5:00 pm ET and Friday from 9:00 am to 12:30 pm ET. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, or by fax.
- Complete and submit the report online
- Regular Mail or Fax: download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the US Food and Drug Administration (FDA).
Company Contact Information
Consumers:
- Allen Fields, Senior Vice President, Clinical and Regulatory Affairs
- 1-800-541-4802
Product Photos:
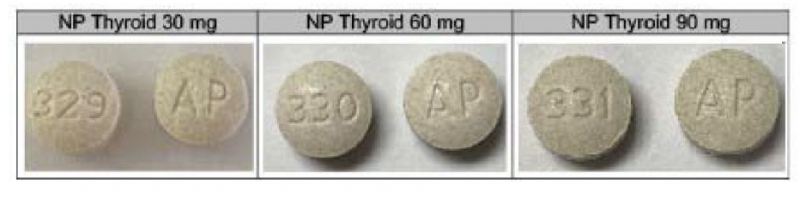

True Metrix® Air Blood Glucose Meter
Date: 4/21/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Trividia Health, Inc. has posted a recall of one (1) True Metrix® Air Blood Glucose Meter with serial number TA1548753. The recalled meter was packaged into a True Metrix Blood Glucose Meter Kit with the lot number and UDI number shown below, and this kit was distributed nationwide in the United States in February 2019.
About this Recall:
Trividia Health, Inc. has announced a nationwide voluntary recall of one (1) True Metrix® Air Blood Glucose Meter distributed in the United States to AssuraMed. The one (1) impacted meter was packaged into a True Metrix Blood Glucose Meter Kit with the Lot number KW0135 and with UDI number (01)00021292006075(17) 200831(10)KW0135(21)TA1548753.
What this means to you:
The recalled meter has an incorrect factory-set unit of measure and displays blood glucose results in mmol/L rather than mg/dL. If this incorrect unit of measure is not noticed, the potential exists for the meter glucose result to be read as a lower blood glucose reading than expected. This can lead to the patient’s glucose level remaining high, which could result in serious injury or lead to life-threatening complications.
Consumers who have or may have the recalled True Metrix Air meter are instructed by the manufacturer to do the following:
- Check to confirm if you have the impacted True Metrix Air meter by obtaining the meter serial number (TA1548753) from the serial number label located on the back of the meter, and the call Trividia Health toll-free at 1-800-518-5726, Monday to Friday, 8 AM to 8 PM EST.
- If you have the impacted True Metrix Air meter, please stop using the meter and call Trividia Health immediately to verify the serial number, and to expedite return and replacement of the recalled True Metrix Air meter at no charge.
- Consumers may continue to test blood glucose using any other Trividia Health blood glucose meter not included in this voluntary recall while waiting for their replacement meter to arrive. Use only the test strips that are intended for use with your blood glucose meter.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Trividia Health, Inc. Issues Nationwide Voluntary Recall for an Isolated True Metrix® Air Blood Glucose Meter with Serial Number TA1548753
FDA Publish Date: 4/21/2020
Trividia Health, Inc. today announced it is initiating a nationwide voluntary recall of one (1) True Metrix® Air Blood Glucose Meter distributed in the United States to one customer (AssuraMed). The meter was not packaged into store-brand or retail branded packaging.
The company has determined that one (1) isolated True Metrix Air Blood Glucose Meter was packaged into a True Metrix Blood Glucose Meter kit and has an incorrect factory-set unit of measure; the meter displays glucose results in mmol/L rather than mg/dL. If a consumer does not notice the incorrect unit of measure, it is possible that the meter glucose result will be read as a lower blood glucose result than expected, and this may result in the patient’s glucose level remaining high, which can lead to serious injury or impairment with risk of death.
There is one (1) affected True Metrix Air meter with serial number TA1548753 that was packaged into a True Metrix Blood Glucose Meter Kit with the Lot number KW0135 and with UDI number (01)00021292006075(17)200831(10)KW0135(21)TA1548753. It has been determined that this kit was distributed nationwide in the United States (US) in February 2019. Trividia Health has not received any reports of patient injury or an adverse event related to this voluntary recall.
Consumers who have or may have the recalled True Metrix Air meter are instructed by the manufacturer to do the following:
-
Check to confirm if you have the affected True Metrix Air meter by obtaining the meter serial number ( TA1548753) from the serial number label located on the back of the meter, and then call Trividia Health toll-free at 1-800-518-5726, Monday – Friday, 8 AM – 8 PM EST.
- If you have the affected True Metrix Air meter, please stop using the meter and call Trividia Health immediately to verify the serial number, and to expedite return and replacement of the affected True Metrix Air meter at no charge.
Consumers may continue to test blood glucose using any other Trividia Health blood glucose meter not included in this voluntary recall while waiting for their replacement meter to arrive. Use only the test strips that are intended for use with your blood glucose meter.
Notifications will be sent to US pharmacies, durable medical equipment providers and distributors who may have received this device from Trividia’s customer.
Patient safety is a top priority at Trividia Health. The company has notified the US Food and Drug Administration (FDA) of this voluntary product recall and is working with the customer who received the affected meter to quickly resolve this matter.
Users within the US may report adverse reactions or quality problems experienced with the use of this product to the FDA’s MedWatch Adverse Event Reporting program either online or by phone.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the US FDA.
Company Contact Information
Consumers: Trividia Health
- 1-800-518-5726
Media: Annmarie Ramos, Sr. Manager, Product Marketing
- 800-342-7226, ext. 3190
Tetracycline HCl Capsules 250 mg and 500 mg
Date: 4/16/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Avet Pharmaceuticals has posted a lot recall of Tetracycline HCl capsules.
About this Recall:
Avet Pharmaceuticals Inc. is initiating a voluntary recall of 8 lots of Tetracycline HCl Capsules USP, 250 mg and 500 mg, 100-count bottles to the consumer/user level. A list of impacted lots is available at the FDA Recall Notification website below.
What this means to you:
Avet is notifying its distributors and customers and is arranging for the return of all recalled products. Consumers should contact their doctor for further guidance and potential change of treatment before they stop taking this drug product.
Consumers with questions regarding this recall should contact Qualanex at 1-888-424-4341 Monday – Friday, 8:00 am – 5:00 pm, EST or via email at recall@qualanex.com. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Avet Pharmaceuticals Inc. Issues Voluntary Nationwide Recall of Tetracycline HCl Capsules USP, 250 mg and 500 mg Due to Failed Dissolution Specifications
FDA Publish Date: 4/16/2020
Avet Pharmaceuticals Inc. (“Avet”), based in East Brunswick, New Jersey, is initiating a voluntary recall of the following lots of Tetracycline HCl Capsules USP, 250 mg and 500 mg, 100-count bottles listed in the table below to the consumer/user level.
|
Product |
NDC Number |
Lot No. |
Expiry Date |
|
Tetracycline HCl Capsules 250 mg, 100 count |
23155-0017-01 |
H190666 |
JUL 2022 |
|
Tetracycline HCl Capsules 500 mg, 100 count |
23155-0018-01 |
G190609 |
JUN 2022 |
|
Tetracycline HCl Capsules 500 mg, 100 count |
23155-0018-01 |
G190610 |
JUN 2022 |
|
Tetracycline HCl Capsules 500 mg, 100 count |
23155-0018-01 |
G190611 |
JUN 2022 |
|
Tetracycline HCl Capsules 500 mg, 100 count |
23155-0018-01 |
L191027 |
NOV 2022 |
|
Tetracycline HCl Capsules 500 mg, 100 count |
23155-0018-01 |
L191028 |
NOV 2022 |
|
Tetracycline HCl Capsules 500 mg, 100 count |
23155-0018-01 |
K190953 |
OCT 2022 |
|
Tetracycline HCl Capsules 500 mg, 100 count |
23155-0018-01 |
K190952 |
OCT 2022 |
These drug products are manufactured by Avet Pharmaceuticals and distributed under the Heritage Pharmaceuticals label. The voluntary recall is being initiated due to low out of specification dissolution test results.
Low dissolution results in less tetracycline available in the body to fight infection. This can lead to treatment failures. For patients with compromised immune systems and the elderly, who may be taking tetracycline to treat a serious infection such as pneumonia, there is a reasonable probability that if there is not enough tetracycline in
2—0415-2020 Class 1 Tetracycline HCl Capsules – Avet Pharmaceuticals Voluntary Nationwide Recall
the body to fight the infection, this could result in rapid progression of the infection and death. To date, Avet has not received adverse event reports or complaints related to this event.
Tetracycline HCl Capsules USP, 250 mg and 500 mg are indicated in the treatment of infections caused by susceptible strains of the designated organisms, including upper and lower respiratory infections, skin and soft tissues infections, infections caused by Rickettsiae, and as an adjunctive therapy in severe acne. The recalled Tetracycline HCl Capsules 250 mg and 500 mg lots were distributed to wholesalers and distributors nationwide in the US between August 2019 and March 2020.
Avet is notifying its distributors and customers and is arranging for the return of all recalled products. Consumers should contact their doctor for further guidance and potential change of treatment before they stop taking this drug product. Pharmacies and healthcare facilities that have the drug product subject to this recall should immediately stop dispensing this drug product.
Consumers with questions regarding this recall should contact Qualanex at 1-888-424-4341, Monday – Friday, 8:00 am – 5:00 pm, EST or via email at recall@qualanex.com. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail or by fax.
- Complete and submit the report online
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the US Food and Drug Administration.
Company Contact Information
Consumers: Avet Pharmaceuticals Inc.
- 1-888-424-4341
- recall@qualanex.com
Tetracycline HCl Capsules 250 mg and 500 mg
Date: 4/16/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Avet Pharmaceuticals has posted a lot recall of Tetracycline HCl capsules.
About this Recall:
Avet Pharmaceuticals Inc. is initiating a voluntary recall of 8 lots of Tetracycline HCl Capsules USP, 250 mg and 500 mg, 100-count bottles to the consumer/user level. A list of impacted lots is available at the FDA Recall Notification website below.
What this means to you:
Avet is notifying its distributors and customers and is arranging for the return of all recalled products. Consumers should contact their doctor for further guidance and potential change of treatment before they stop taking this drug product.
Consumers with questions regarding this recall should contact Qualanex at 1-888-424-4341 Monday – Friday, 8:00 am – 5:00 pm, EST or via email at recall@qualanex.com. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Avet Pharmaceuticals Inc. Issues Voluntary Nationwide Recall of Tetracycline HCl Capsules USP, 250 mg and 500 mg Due to Failed Dissolution Specifications
FDA Publish Date: 4/16/2020
Avet Pharmaceuticals Inc. (“Avet”), based in East Brunswick, New Jersey, is initiating a voluntary recall of the following lots of Tetracycline HCl Capsules USP, 250 mg and 500 mg, 100-count bottles listed in the table below to the consumer/user level.
|
Product |
NDC Number |
Lot No. |
Expiry Date |
|
Tetracycline HCl Capsules 250 mg, 100 count |
23155-0017-01 |
H190666 |
JUL 2022 |
|
Tetracycline HCl Capsules 500 mg, 100 count |
23155-0018-01 |
G190609 |
JUN 2022 |
|
Tetracycline HCl Capsules 500 mg, 100 count |
23155-0018-01 |
G190610 |
JUN 2022 |
|
Tetracycline HCl Capsules 500 mg, 100 count |
23155-0018-01 |
G190611 |
JUN 2022 |
|
Tetracycline HCl Capsules 500 mg, 100 count |
23155-0018-01 |
L191027 |
NOV 2022 |
|
Tetracycline HCl Capsules 500 mg, 100 count |
23155-0018-01 |
L191028 |
NOV 2022 |
|
Tetracycline HCl Capsules 500 mg, 100 count |
23155-0018-01 |
K190953 |
OCT 2022 |
|
Tetracycline HCl Capsules 500 mg, 100 count |
23155-0018-01 |
K190952 |
OCT 2022 |
These drug products are manufactured by Avet Pharmaceuticals and distributed under the Heritage Pharmaceuticals label. The voluntary recall is being initiated due to low out of specification dissolution test results.
Low dissolution results in less tetracycline available in the body to fight infection. This can lead to treatment failures. For patients with compromised immune systems and the elderly, who may be taking tetracycline to treat a serious infection such as pneumonia, there is a reasonable probability that if there is not enough tetracycline in the body to fight the infection, this could result in rapid progression of the infection and death. To date, Avet has not received adverse event reports or complaints related to this event.
Tetracycline HCl Capsules USP, 250 mg and 500 mg are indicated in the treatment of infections caused by susceptible strains of the designated organisms, including upper and lower respiratory infections, skin and soft tissues infections, infections caused by Rickettsiae, and as an adjunctive therapy in severe acne. The recalled Tetracycline HCl Capsules 250 mg and 500 mg lots were distributed to wholesalers and distributors nationwide in the US between August 2019 and March 2020.
Avet is notifying its distributors and customers and is arranging for the return of all recalled products. Consumers should contact their doctor for further guidance and potential change of treatment before they stop taking this drug product. Pharmacies and healthcare facilities that have the drug product subject to this recall should immediately stop dispensing this drug product.
Consumers with questions regarding this recall should contact Qualanex at 1-888-424-4341, Monday – Friday, 8:00 am – 5:00 pm, EST or via email at recall@qualanex.com. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the US Food and Drug Administration.
Company Contact Information
Consumers: Avet Pharmaceuticals Inc.
- 1-888-424-4341
- recall@qualanex.com
Nizatidine Oral Solution 15 mg/mL
Date: 4/15/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall information.
Amneal Pharmaceuticals has posted a lot recall of Nizatidine Oral Solution 15 mg/mL.
About this Recall:
Amneal Pharmaceuticals, LLC, Bridgewater, New Jersey is voluntarily recalling three lots of Nizatidine Oral Solution, 15 mg/mL (75 mg/5mL), packaged in 480 mL bottles to the Consumer Level. Nizatidine Oral Solution was distributed by Gemini Laboratories, LLC, a wholly owned subsidiary of Amneal Pharmaceuticals. The three recalled lots are identified in the FDA Recall Notification link below. Nizatidine Oral Solution is being recalled due to potential N-Nitrosodimethylamine (NDMA) amounts exceeding the levels established by the FDA.
What this means to you:
Customers who purchased the impacted product directly from Amneal can call Inmar at (855) 319-4807, Monday – Friday, 8:00 am – 6:00 pm, EST, or e-mail at DrugSafety@amneal.com for further information. Consumers should contact their physician or other healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Consumers who have Nizatidine Oral Solution which is being recalled should stop using the product and can call Inmar at 855-319-4807, Monday – Friday, 8:00 am – 5:00 pm, EST for further information.
Consumers who would like to report adverse reactions or quality problems experienced with the use of this product can contact Amneal Drug Safety by phone at 1-877-835-5472, Monday thru Friday, 8:00 am – 6:00 pm, EST, or e-mail at DrugSafety@amneal.com.
Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to the use of this drug product.
For more information regarding this FDA Recall Notification, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Amneal Pharmaceuticals, LLC. Issues Voluntary Nationwide Recall of Nizatidine Oral Solution, 15 mg/mL, Due to Potential Levels of NDMA Impurity Amounts Above the Levels Established by FDA
FDA Publish Date: 4/15/2020
Amneal Pharmaceuticals, LLC, Bridgewater, New Jersey is voluntarily recalling three lots of Nizatidine Oral Solution, 15 mg/mL (75 mg/5mL), packaged in 480 mL bottles to the Consumer Level. Nizatidine Oral Solution was distributed by Gemini Laboratories, LLC, a wholly owned subsidiary of Amneal Pharmaceuticals. The three recalled lots are identified in the table below. Nizatidine Oral Solution is being recalled due to potential N-Nitrosodimethylamine (NDMA) amounts exceeding the levels established by the FDA.
Risk Statement: NDMA is classified as a probable human carcinogen (a substance that could cause cancer) based on results from laboratory tests. NDMA is a known environmental contaminant and found in water and foods, including meats, dairy products, and vegetables.
Amneal Pharmaceuticals, LLC has not received any reports of adverse events that have been confirmed to be directly related to this recall. Nizatidine Oral Solution manufactured by Amneal, is a prescription oral product used for the short-term treatment and maintenance therapy of ulcers and for the treatment of esophagitis and associated heartburn due to gastroesophageal reflux disease (GERD).
The Nizatidine Oral Solution lots subject to the recall can be identified by the NDC number and lot number listed on the product label:
|
NDC No. |
Description |
Lot |
Expiration Date |
|
60846-0301-15 |
Nizatidine Oral Solution |
06598004A |
04/2020 |
|
60846-0301-15 |
Nizatidine Oral Solution |
06599001A |
12/2020 |
|
60846-0301-15 |
Nizatidine Oral Solution |
06599002A |
12/2020 |
The affected Nizatidine Oral Solution lots were distributed directly to wholesalers who further distributed to retail pharmacies and consumers nationwide in the USA.
Amneal is notifying its direct customers by mailing (FED Ex Standard Overnight) a recall notification letter and is arranging for return of all recalled product. Anyone with an existing inventory of the product should quarantine the recalled lots immediately.
Customers who purchased the impacted product directly from Amneal can call Inmar at (855) 319-4807, Monday – Friday, 8:00 am – 6:00 pm, EST, or e-mail at DrugSafety@amneal.com for further information. Consumers should contact their physician or other healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Consumers who have Nizatidine Oral Solution which is being recalled should stop using the product and can call Inmar at 855-319-4807, Monday – Friday, 8:00 am – 5:00 pm, EST for further information.
Consumers who would like to report adverse reactions or quality problems experienced with the use of this product can contact Amneal Drug Safety by phone at 1-877-835-5472, Monday thru Friday, 8:00 am – 6:00 pm, EST, or e-mail at DrugSafety@amneal.com.
Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to the use of this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail or by fax.
- Complete and submit the report online
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the U.S. Food and Drug Administration.
Company Contact Information
Consumers: Inmar
- 855-319-4807
- DrugSafety@amneal.com
Media: Ms. Candis Edwards
Ranitidine Products
Date: 4/1/2020
At Magellan Rx Management, we want to help you get the best possible care. We have created a site to share drug recall and withdrawal information. The FDA has posted a full market withdrawal of ALL Ranitidine Products.
About this Withdrawal:
The U.S. Food and Drug Administration (FDA) today announced it is requesting manufacturers withdraw all prescription and over-the-counter (OTC) ranitidine drugs from the market immediately. This is the latest step in an ongoing investigation of a contaminant known as NNitrosodimethylamine (NDMA) in ranitidine medications (commonly known by the brand name Zantac).
What this means to you:
Patients and consumers should contact their physician or health care provider with any questions related to this withdrawal and/or if they have experienced any problems that may be related to taking Ranitidine Products. The FDA recommends consumers stop taking OTC ranitidine drugs and consider other approved OTC products if they wish to continue treating their condition. The FDA also recommends if you have had a prescription for this drug, to talk with your doctor or pharmacist before stopping this drug. Because of the current COVID-19 pandemic, the FDA recommends consumers not take their drugs to a drug take-back location but follow the specific disposal directions in the medication guide or package insert or follow the FDA’s recommended steps, which include ways to safely dispose of these drugs at home.
For more information regarding this FDA Withdrawal Notification, please refer to the FDA website.
Complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2
FDA Requests Removal of All Ranitidine Products (Zantac) from the Market FDA
Publish Date: 04/01/2020
The U.S. Food and Drug Administration today announced it is requesting manufacturers withdraw all prescription and over-the-counter (OTC) ranitidine drugs from the market immediately. This is the latest step in an ongoing investigation of a contaminant known as N-Nitrosodimethylamine (NDMA) in ranitidine medications (commonly known by the brand name Zantac). The agency has determined that the impurity in some ranitidine products increases over time and when stored at higher than room temperatures and may result in consumer exposure to unacceptable levels of this impurity. As a result of this immediate market withdrawal request, ranitidine products will not be available for new or existing prescriptions or OTC use in the U.S.
“The FDA is committed to ensuring that the medicines Americans take are safe and effective. We make every effort to investigate potential health risks and provide our recommendations to the public based on the best available science. We didn’t observe unacceptable levels of NDMA in many of the samples that we tested. However, since we don’t know how or for how long the product might have been stored, we decided that it should not be available to consumers and patients unless its quality can be assured,” said Janet Woodcock, M.D., director of the FDA’s Center for Drug Evaluation and Research. “The FDA will continue our efforts to ensure impurities in other drugs do not exceed acceptable limits so that patients can continue taking medicines without concern.”
NDMA is a probable human carcinogen (a substance that could cause cancer). In the summer of 2019, the FDA became aware of independent laboratory testing that found NDMA in ranitidine. Low levels of NDMA are commonly ingested in the diet, for example NDMA is present in foods and in water. These low levels would not be expected to lead to an increase in the risk of cancer. However, sustained higher levels of exposure may increase the risk of cancer in humans. The FDA conducted thorough laboratory tests and found NDMA in ranitidine at low levels. At the time, the agency did not have enough scientific evidence to recommend whether individuals should continue or stop taking ranitidine medicines, and continued its investigation and warned the public in September 2019 of the potential risks and to consider alternative OTC and prescription treatments.
New FDA testing and evaluation prompted by information from third-party laboratories confirmed that NDMA levels increase in ranitidine even under normal storage conditions, and NDMA has been found to increase significantly in samples stored at higher temperatures, including temperatures the product may be exposed to during distribution and handling by consumers. The testing also showed that the older a ranitidine product is, or the longer the length of time since it was manufactured, the greater the level of NDMA. These conditions may raise the level of NDMA in the ranitidine product above the acceptable daily intake limit.
With today’s announcement, the FDA is sending letters to all manufacturers of ranitidine requesting they withdraw their products from the market. The FDA is also advising consumers taking OTC ranitidine to stop taking any tablets or liquid they currently have, dispose of them properly and not buy more; for those who wish to continue treating their condition, they should consider using other approved OTC products. Patients taking prescription ranitidine should speak with their health care professional about other treatment options before stopping the medicine, as there are multiple drugs approved for the same or similar uses as ranitidine that do not carry the same risks from NDMA. To date, the FDA’s testing has not found NDMA in famotidine (Pepcid), cimetidine (Tagamet), esomeprazole (Nexium), lansoprazole (Prevacid) or omeprazole (Prilosec).
In light of the current COVID-19 pandemic, the FDA recommends patients and consumers not take their medicines to a drug take-back location but follow the specific disposal instructions in the medication guide or package insert or follow the agency’s recommended steps, which include ways to safely dispose of these medications at home.
The FDA continues its ongoing review, surveillance, compliance and pharmaceutical quality efforts across every product area, and will continue to work with drug manufacturers to ensure safe, effective and high-quality drugs for the American public. The FDA encourages health care professionals and patients to report adverse reactions or quality problems with any human drugs to the agency’s MedWatch Adverse Event Reporting program:
- Complete and submit the report online at www.fda.gov/medwatch/report.htm; or
- Download and complete the form, then submit it via fax at 1-800-FDA-0178.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices.
The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
Inquiries Media: Consumer: Sarah Peddicord
- 888-INFO-FDA
- 301-796-2805
Related Information
- Questions and Answers: NDMA impurities in ranitidine (commonly known as Zantac)
- What to Know and Do About Possible Nitrosamines in Your Medication
- Information about Nitrosamine Impurities in Medications Link to full FDA Alert: FDA Alert Ranitidine HCL Full Market Withdrawal
Link to full FDA Alert: FDA Alert Ranitidine HCL Full Market Withdrawal
Ketorolac Tromethamine Injection, 30mg/mL
FDA Publish Date: 03/05/2020
At Magellan Rx Management, we want to help you get the best possible care. We have a website to share drug recall information.
Hikma Pharmaceuticals has posted a lot recall of Ketorolac Tromethamine Injection, 30mg/mL.
About this Recall:
Hikma Pharmaceuticals is voluntarily extending its prior recall of certain lots of Ketorolac Tromethamine Injection USP 30mg/mL. The drug is being recalled because small black pieces, were seen in some of the recalled lots.
What this means to you:
Patients and consumers should contact their physician or health care provider if they have experienced any problems that may be related to taking or using Ketorolac Tromethamine Injection, 30mg/mL.
For more information regarding FDA Recall Notifications, please refer to the FDA website.
FDA contact information for reporting adverse events/quality complaints can be reached online or by calling the FDA at 1-888-INFO-FDA (1-888-463-6332) and then select prompt #2.
Hikma Pharmaceuticals USA Inc. Extends Voluntary Nationwide Recall of Ketorolac Tromethamine Injection, USP 30mg/mL, 1mL Fill/2mL Vials Due to the Potential Presence of Small Particulates
FDA Publish Date: 03/05/2020
Hikma Pharmaceuticals PLC (Hikma, Group), today announces that its subsidiary Hikma Pharmaceuticals USA Inc. (formerly known as West-Ward Pharmaceuticals Corp.; “Hikma”) is voluntarily extending its previously-announced recall of certain lots (listed below) of Ketorolac Tromethamine Injection USP 30mg/mL, 1mL fill/2mL vials to the medical facility and retail levels.
The product is being recalled due to the presence of small visible particulate matters of a gelatinous/oily nature that appear black in some of the recalled lots. On December 23, 2019, Hikma voluntarily initiated a recall of this product to the direct customer level.
In coordination with the U.S. Food and Drug Administration (FDA), Hikma is extending the recall to the medical facility and retail levels. Administration of the affected product could potentially result in the deposition of particulates in the lungs of patients, which could result in multiple pulmonary microemboli with subsequent acute respiratory distress for patients receiving the drug intravenously.
Although Hikma has not received any reports of adverse events related to this issue, it is nonetheless extending its recall of these products out of an abundance of caution and to promote patient safety, which is Hikma’s highest priority.
The lots being recalled were manufactured between March 22, 2018, and February 21, 2019. Hikma investigated the cause of the problem and decided to suspend manufacturing of this product until an appropriate solution can be implemented to prevent recurrence.
Ketorolac Tromethamine Injection is a nonsteroidal anti-inflammatory drug (“NSAID”) that is indicated for the shortterm (up to 5 days in adults) management of moderately severe acute pain that requires analgesia at the opioid level.
The affected lot numbers and expiration dates being recalled are as follows:

The product can be identified by name and NDC and lot codes, which are clearly stated on the product label, along with Hikma Pharmaceuticals USA Inc./West-Ward Pharmaceutical Corp.’s name and address. Images of the vial and shelf pack labels are included below:
The product was distributed to Hikma’s direct customers nationwide. Hikma notified its direct customers as part of the initial recall, asking them to contact medical and retail level facilities to locate and remove the recalled product from distribution channels and return the recalled lots to Hikma. Hikma is now asking customers at the medical and retail level facilities to locate and remove the recalled product from their channels and return the recalled lots to Hikma.
For recall inquiries, please contact Hikma using the information provided below:

Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product. Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail or by fax.
- Complete and submit the report online
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the U.S. Food and Drug Administration. Link to FDA website.
Click here for language services (translation, TTY, etc.)